|
在分子生物学研究中,DNA的序列分析是进一步研究和改造目的基因的基础。目前用于测序的技术主要有Sanger等(1977)发明的双脱氧链末端终止法和Maxam和Gilbert(1977)发明的化学降解法。这二种方法在原理上差异很大,但都是根据核苷酸在某一固定的点开始,随机在某一个特定的碱基处终止,产生A,T,C,G四组不同长度的一系列核苷酸,然后在尿素变性的PAGE胶上电泳进行检测,从而获得DNA序列。目前Sanger测序法得到了广泛的应用。 Sanger法测序的原理就是利用一种DNA聚合酶来延伸结合在待定序列模板上的引物。直到掺入一种链终止核苷酸为止。每一次序列测定由一套四个单独的反应构成,每个反应含有所有四种脱氧核苷酸三磷酸(dNTP),并混入限量的一种不同的双脱氧核苷三磷(ddNTP)。由于ddNTP缺乏延伸所需要的3-OH基团,使延长的寡聚核苷酸选择性地在G、A、T或C处终止。终止点由反应中相应的双脱氧而定。每一种dNTPs和ddNTPs的相对浓度可以调整,使反应得到一组长几百至几千碱基的链终止产物。它们具有共同的起始点,但终止在不同的的核苷酸上,可通过高分辨率变性凝胶电泳分离大小不同的片段,凝胶处理后可用X-光胶片放射自显影或非同位素标记进行检测。 一、非同位素银染测序系统操作技术概述 Promega公司的SILVER SEQUENCETM DNA测序系统是一种无放射性的序列分析系统,它通过灵敏的银染方法检测凝胶中的条带。 银染提供了一种对于放射性或荧光法来说更加快速,廉价的替代方法。测序结果可以在同一天内得到;电泳完成后经90分钟就可读序,这是常规的放射性测序法做不到的。 此外,SILVER SEQUENCETM系统用未修饰的5'OH寡聚核苷酸作为引物,减少了特殊修饰寡聚核苷酸的花费。该系统不需要放射性方法中对同位素的谨慎操作,也不需要荧光法或化学发光技术的昂贵试剂。另外,也不需要象大多数荧光法那样用仪器来检测序列条带。 Taq DNA聚合酶在95℃时极强的热稳定性。本系统利用的测序级Taq DNA聚合酶是一种Taq DNA聚合酶的修饰产品,对于双链DNA模板有非常好的效果,具有高度的准确性,能产生均一的条带,且背景低。 SILVER SEQUENCETM系统包含被修饰的核苷酸混合物,如7-去氮dGTP(7-deaza dGTP,或dITP)替代dGTP可清除由GC丰富区域所引起的条带压缩现象。 退火温度是热循环测序中最重要的因素。高退火温度可减少模板二级结构。提高引物结合模板配对的严谨性。链重退火和模板二级结构则限制了小片断PCR产物(<500bp)得到清楚的序列数据的能力。引物延伸起始于每个循环的退火阶段。在较低温度时,聚合酶可能会遇到坚固的二级结构区域,它可导致聚合酶解离。则在四个电泳道中均有同一相对位置的条带。因为这些原因,应该使用尽可能高的退火温度。对于有牢固二级结构的模板建议使用95℃变性、70℃退火/延伸的循环模式。一般来说,较长的引物及GC含量高的引物能得到较强的信号。实验结果表明,>24mer的GC含量约为50%的引物可得到最佳结果。 由于本系统采用热循环装置, 与常规的测序方法相比具有如下几点好处:(1).本方法线性扩增模板DNA产生足够的产物使银染技术能够检测序列条带,测序反应需要0.03~2pmol模板DNA,随模板种类而定。(2). 在每一个变性循环中的高温可以取代对双链DNA(dsDNA)模板的碱变性及乙醇沉淀过程,变性循环也有助于消除由于线性dsDNA模板(如PCR反应产物)快速重退火所引起的问题。(3). 高温聚合酶反应减弱了DNA模板的二级结构,允许聚合酶穿过高度二级结构化的区域。 二、材料 待测已提纯的DNA,可为单链,也可为双链。 三、设备 高压电泳仪,测序用电泳槽,制胶设备,PCR仪。 四、试剂 (1)SILVER SEQUENCETM DNA测序试剂盒。 (2)丙烯酰胺和甲叉双丙烯酰胺储备液(38%丙烯酰胺 W/V,2%甲叉双丙烯酰胺 W/V):95g丙烯酰胺,5g甲叉双丙烯酰胺溶于140ml 双蒸水中,定容至250ml,0.45mm过滤器过滤后,贮于棕色瓶中,置于4℃冰箱可保存2周。 (3)10%过硫酸铵,0.5g过硫酸铵溶于4ml水中,定容至5ml,应新配新用。 (4)10×TBE缓冲液(1 mol/L Tris, 0.83mol/L 硼酸,10mmol/L EDTA): 121.1g Tris,51.35g硼酸,3.72g Na2EDTA ·2H2O,溶于双蒸水中定容至1升,置于4℃下可贮存2周,其pH约为8.3。 (5)TBE电极缓冲液:10×TBE 缓冲液稀释至1×TBE备用。 (6)TEMED (7)固定/停止溶液:10%冰醋酸(V/V)配制2升备用。 (8)染色溶液:硝酸银2克,甲醛3ml,溶于2升超纯水中备用。 (9)显影溶液:60克碳酸钠(Na2CO3)溶于2升超纯水中,使用前加3ml 37%甲醛和40ml硫代硫酸钠溶液(10mg/ml)。 (10)95%乙醇。 (11)0.5%冰乙酸。 (12)Sigmacote (Sigma CAT. #SL-2)。 |
|
五、操作步骤 成功地使用银染测序系统需要对提供的操作方法进行仔细考虑。银染不如放射性检测法灵敏,而需要更多的模板量,此外,也不可能通过延长X光胶片曝光时间的方法增加信号强度。因此,请使用推荐的DNA模板量, 每次均使用所提供的对照检查系统的可靠性,并且注意如下几点: (1) DNA的浓度和纯度必须经过琼脂糖凝胶电泳或荧光法测定,样品应与已知量DNA一起电泳。 (2) 分光光度法对于很多DNA提取物包括质粒小量制备来说,并不能给出一个可信的DNA浓度估计,混杂的染色体DNA、蛋白、RNA、有机物及无机化合物均可能有260nm光吸收。因此,分光光度法常常错误地高估DNA浓度。 (3) DNA制备过程中用核糖核酸酶处理所产生核糖核苷酸,虽然它们在电泳后DNA样品的前面,并不能观察到,但它们仍会有260nm光吸收。 (一)测序反应 1. 对于每组测序反应,标记四个0.5ml eppendorf管(G、A、T、C)。每管加入2ml适当的d/ddNTP混合物(d/ddNTP Mix)。各加入1滴(约20ul)矿物油,盖上盖子保存于冰上或4℃备用。 2. 对于每组四个测序反应,在一个eppendorf管中混合以下试剂: 3. 在引物/模板混合物(以上第2步)中加入1.0ml测序级Taq DNA聚合酶(5u/ml)。用吸液器吸动几次混匀。 4. 从第3步的酶/引物/模板混合物中吸取4ml加入每一个d/ddNTP混合物的管内。 5. 在微量离心机中离心一下,使所有的溶液位于eppendorf管底部。 6. 把反应管放入预热至95℃的热循环仪,以【注意】中循环模式为基准,开始循环程序。对于每个引物/模板组合都必须选择最佳退火温度。下列程序一般能读出从引物开始350碱基的长度。 7. 热循环程序完成后,在每个小管内加入3μlDNA测序终止溶液,在微量离心机中略一旋转,终止反应。 【注意】 1、测序所用模板DNA的量一般按下面要求加入: 2、计算与4.5pmol相当的引物纳克数可用以下一般公式: 3、为阻止Taq DNA聚合酶延伸非特异性退火引物, 热循环仪必须预热至95℃。温度变换应越快越好。下面的循环时间不包括变温时间。如果你无法确定使用何种模式,建议从模式1开始。 4. 在加入终止溶液之后样品可在4℃保存过夜。 |
|
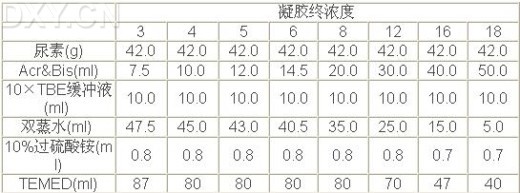
(二)测序凝胶板的制备 1、玻璃板的处理: 银染测序的玻璃板一定要非常清洁,一般先用温水和去污剂洗涤,再用去离子水冲洗玻璃板,除去残留的去污剂,最后用乙醇清洗玻璃板。玻璃板上遗留的去污剂微膜可能导致凝胶染色时背景偏高(棕色)。短玻璃板经粘合溶液处理可将凝胶化学交联于玻璃板上。这一步对于在银染操作过程中防止凝胶撕裂至关重要。 (1)短玻璃板的处理 【注意】 (2)长玻璃板的处理: 【注意】 1. 用过的凝胶可在水中浸泡后用剃须刀片或塑料刮刀刮去。玻璃板须用去污剂完全清洗。或者凝胶用10%NaOH浸泡后除去。为防止交叉污染,用于清洗短玻璃板的工具必须与清洗长玻璃板的工具分开,如果出现交叉污染,以后制备的凝胶可能撕裂或变得松驰。 2、凝胶的制备: (1)玻璃板经粘合硅胶和Sigmacote处理后,即可固定玻璃板。该方法是用0.2mm或0.4mm厚的边条置于玻璃板左右两侧,将另一块玻璃板压于其上。在长玻璃板的一侧插入鲨鱼齿梳平的一面边缘,用夹子固定住。 (2)根据所需要的凝胶浓度,按下表制备测序凝胶,一般6%~8%的胶浓度可获得较好的结果。配制过程中,先用适量双蒸水溶解尿素,再加入Acr&Bis和10×TBE缓冲液,再用双蒸水调终体积至99.2ml,并用0.45mm的滤膜过滤,然后加过硫酸铵和TEMED。溶解尿素时不必加热。如果确需加热则应等溶液完全冷却后,方可加入TEMED和过硫酸铵。一般在胶灌制后4~6分钟,即开始聚合,如果聚合不好,则应使用高浓度的TEMED和过硫酸铵。
(3)胶配制好后,即可灌制胶板。一般是将凝胶沿着压条边缘缓慢地倒入玻璃板的槽中,倒完后,静止放置使之聚合完全。 【注意】 1、使用夹子固定玻璃板时,最好夹子的力量稍大一些,防止因力量不足使灌胶的过程中出现漏胶液现象。 2、灌制凝胶的过程中要严防产生气泡,否则影响测序的结果。 (三)电泳 1、预电泳 (1)当凝胶聚合完全后,拨出鲨鱼齿梳,将该梳子反过来,把有齿的一头插入凝胶中,形成加样孔。 (2)立即将胶板固定在测序凝胶槽中,一般测序凝胶槽的上下槽是分开的,因而只有在固定好凝胶板后,方能加入TBE缓冲液。 (3)稀释10×TBE缓冲液至1×TBE,将该缓冲液加入上下二个电泳槽中,去除产生的气泡,接上电源准备预电泳。 (4)有些电泳槽,如LKB的Macrophor等是使用水浴加热的,则应先将水浴加热至55℃后进行预电泳。有的不使用水浴加热,依靠电泳过程中自身产生的热进行保温,如上海求精有机玻璃仪器生产的测序电泳槽,这种槽需夹上二块散热铝板,使整个凝胶板的温度一致。 (5)按30V/cm的电压预电泳20~30分钟。预电泳的过程是去除凝胶的杂质离子,同时使凝胶板达到所需的温度。高温电泳可防止GC丰富区形成的发夹状结构,影响测序的结果。 【注意】 (1)用鲨鱼齿梳制作加样孔时,应注意将齿尖插入胶中0.5mm左右,千万注意不能使加样孔渗漏,否则得不到正确的结果。 (2)应时刻注意上面电泳槽中的缓冲液是否渗漏,否则极易造成短路而损坏电泳仪。 2、样品的制备: 当在预电泳时,即可进行样品的制备,将反应完毕的样品在沸水浴中加热1~3分钟,立即置于冰上即可。如果样品长时间不用,则应重新处理。可使用4~6%聚丙烯酰胺凝胶,胶厚0.4mm。厚度小于0.4mm的胶可能导致信号太弱。加样时不必吸去上层覆盖的矿物油,但要小心地吸取矿物油下的蓝色样品。 3、上样及电泳 关闭电泳仪,用移液枪吸缓冲液清洗样品孔,去除在预电泳时扩散出来的尿素,然后立即用毛细管进样器吸取样品,加入样品孔中。上样顺序一般为G、A、T、C。加样完毕后,立即电泳。开始可用30V/cm进行电泳,5分钟后可提高至40~60V/cm,并保持恒压状态。一般来说,一个55cm长,0.2mm厚的凝胶板,在2500V恒压状态下电泳2小时即可走到底部,同时在电泳过程中,电流可稳定地从28mA降至25mA。为了能读到更长的序列,可采用两轮或多轮上样。 【注意】 (1)上样电泳时,一定要注意凝胶板的温度是否达到55℃左右,如果还没有达到,则应等温度达到后才能上样电泳。 (2)一般来说电泳时,不宜使用太高的电压,因为太高的电压会使凝胶的分辨率降低,并且使带扩散。电泳中可进行恒功率电泳。 |
|
(四)测序凝胶的银染 染色过程要求凝胶浸在塑料盘中。因而至少使用两个盘子,大小与玻璃板类似。在盘中加入新鲜溶液之前须用高质量的水洗涤盘子。 1. 电泳完毕后用一个塑料片子小心地分开两板,凝胶应该牢固地附着在短玻璃板上。 2. 固定凝胶:将凝胶(连玻璃板)放入塑料盘,用固定/停止溶液浸没,充分振荡20分钟或直至样品中染料完全消失,胶可在固定/停止溶液中保存过夜(不振荡)。保留固定/停止溶液,用于终止显影反应。 3. 洗胶:用超纯水振荡洗胶3次,每次2分钟。从水中取出,当转移至下一溶液时拿着胶板边沿静止10~20秒,使水流尽。 4. 凝胶染色:把凝胶移至染色溶液充分摇动30分钟。 5. 凝胶显影: 6. 固定凝胶:在显影液中直接加入等体积的固定/停止溶液。停止显影反应,固定凝胶。 7. 在超纯水中浸洗凝胶两次,每次2分钟,注意在本操作中戴手套拿着胶板边缘避免在胶上印上指纹。 8. 将凝胶置于室温干燥或用抽气加热法干燥。在可见光灯箱或亮白,黄色背景(如纸)上观察凝胶,若需永久保存的记录, 则可用EDF胶片保留实验结果。 【注意】 测序产物的银染是显现序列信息的一种新方法,本系统的成败受几个因素的影响。 1. 水的质量对于染色的成功极其重要。超纯水(NANOpureR或Milli-QR的水)或双蒸水可获得较好的效果,如果水中有杂质,则低分子量条带可能无法出现。 2. 碳酸钠也非常重要。使用新鲜的,美国化学学会级碳酸钠较好,如Fisher和Kodak ACS试剂级碳酸钠(Fisher Cat #S263-500或S262-3,或Kodak Cat #109-1990),一般可获得较好的结果。 3. 染色后的洗涤步骤是非常关键的。如果凝胶洗涤时间太长,银颗粒会脱离DNA,产生很少或没有序列信号。如果洗涤时间过长,染色步骤可以重新进行。 4. 如果凝胶厚度超过0.4mm或丙烯酰胺浓度高于4~6%,则有必要延长固定和染色的时间。如果凝胶比0.4mm薄,染色反应后的洗涤必须缩短至不超过5秒。 5. 在室温下进行所有步骤,显影反应除外。显影溶液必须预冷至10~12℃以减小背景杂色。注意:临用前在显影溶液中加入甲醛和硫代硫酸钠。用新配的染色及显影溶液。不要重复使用任何溶液。 (五)EDF胶片显影 使用EDF胶片可增强测序条带的对比度,如果测序胶上条带很淡,我们建议把数据转移至EDF胶片,银染胶在其影像转移至EDF胶片之后可增强条带可读性。 1. 在暗室内,将染色过的粘于玻璃板的凝胶(胶面向上)置于荧光灯箱上。如有合适的漫射板,亦可用白灯箱,为确保曝光时间,用一小条EDF胶片曝光不同时间,检查不同的曝光强度,一般曝光20~40秒可得较好结果。 2. 在红灯下找到EDF胶片有缺刻的一角,然后把胶片置于凝胶上,使缺口位于左上角。由于EDF胶片是单面的,因此必须确保缺口在左上角。 3. 在EDF胶片上放置一干净、干燥的玻璃板,开亮灯箱约20秒。 4. 用冲显放射自显影胶片的步骤手工显影EDF胶片,可使用下列操作过程: 【注意】 1. 进行EDF胶片显影之前凝胶必须完全干燥。戴手套进行操作,以免印上指纹。同时注意EDF胶片不能用自动胶片处理器。 2. 对于不同的光源最佳曝光时间可能不同,通过对一小条EDF胶片曝光不同时间以选择你所用光源的最佳曝光时间,参阅胶片说明书。 3. 曝光时间短则EDF胶片影像较深,曝光时间长则有助于减弱背景。 |
→如果您认为本词条还有待完善,请 编辑词条
词条内容仅供参考,如果您需要解决具体问题
(尤其在法律、医学等领域),建议您咨询相关领域专业人士。
0
收藏到: