|
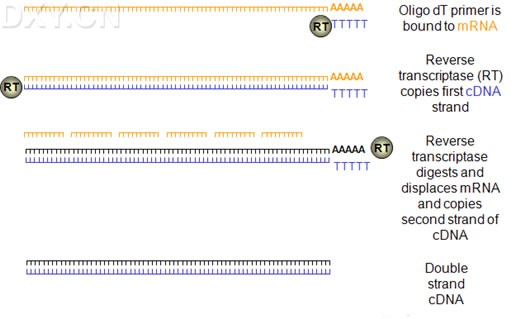
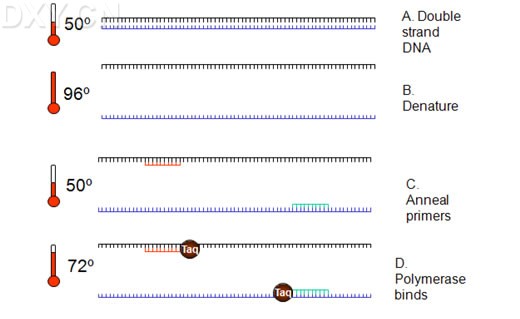
热启动PCR 热启动PCR是除了好的引物设计之外,提高PCR特异性最重要的方法之一。尽管Taq DNA聚合酶的最佳延伸温度在72℃,聚合酶在室温仍然有活性。因此,在进行PCR反应配制过程中,以及在热循环刚开始,保温温度低于退火温度时会产生非特异性的产物。这些非特异性产物一旦形成,就会被有效扩增。在用于引物设计的位点因为遗传元件的定位而受限时,如site-directed突变、表达克隆或用于DNA工程的遗传元件的构建和操作,热启动PCR尤为有效。 限制Taq DNA聚合酶活性的常用方法是在冰上配制PCR反应液,并将其置于预热的PCR仪。这种方法简单便宜,但并不能完成抑制酶的活性,因此并不能完全消除非特异性产物的扩增。 热启动通过抑制一种基本成分延迟DNA合成,直到PCR仪达到变性温度。包括延缓加入Taq DNA聚合酶在内的大部分手工热启动方法十分烦琐,尤其是对高通量应用。其他的热启动方法使用蜡防护层将一种基本成分,如镁离子或酶,包裹起来,或者将反应成分,如模板和缓冲液,物理地隔离开。在热循环时,因蜡熔化而把各种成分释放出来并混合在一起。象手动热启动方法一样,蜡防护层法比较烦琐,易于污染,不适用于于高通量应用。 Platinum DNA聚合酶对于自动热启动PCR来说方便高效。Platinum Taq DNA聚合酶的成分为复合有抗Taq DNA聚合酶单克隆抗体的重组Taq DNA聚合酶。此酶在常温下活性被封闭,要在94℃- 95℃下加热数分钟才能够恢复酶活性。同经化学修饰用于热启动的Taq DNA聚合酶相比,Platinum酶不需要在94℃延时保温(10到15分钟)以激活聚合酶。使用PlatinumTaq DNA聚合酶,在94℃进行2分钟就可以恢复90%的Taq DNA聚合酶活性。 Touch-down PCR Touch-down PCR又称降落PCR.即选定一个温度范围,如50—35 ℃ ,每降1-2 ℃进行1-2个循环,然后在50度下进行15个循环。 Touch-down的原理 随着退火温度的降低,特异性逐步降低,但特异性条带在温度较高时已经扩增出来,其浓度远远超过非特异性条带,随着退火温度的降低,特异性条带优先被扩增。 选择初始复性温度的原则 起始复性温度应该比引物的Tm值高出5-10度,然后每个循环递减1-2度 Touch-down PCR的应用范围 low copy of targeted DNA; high degree degeneracy (or less specific) of the first set of primers; RT-PCR using oligo-dT. RT-PCR RNA的多聚酶反应(RT-PCR)是以RNA 为模板,联合逆转录反应(reverse transcription,RT)与PCR,可用于检测单个细胞或少数细胞中少于10个拷贝的RNA模板。 RNA扩增包括两个步骤 在单引物的介导下和逆转录酶的催化下,合成RNA的互补cDNA;加热后cDNA与RNA链解离,然后与另一引物退火,并由DNA聚合酶催化引物延伸生成双链靶DNA, 最后扩增靶DNA。
cDNA第二链的合成方法有以下几种: 1、自身引导法 合成的单链cDNA 3'端能够形成一短的发夹结构,这就为第二链的合成提供了现成的引物,当第一链合成反应产物的DNA:RNA杂交链变性后利用大肠杆菌DNA聚合酶Ⅰ Klenow片段或反转录酶合成cDNA第二链,最后用对单链特异性的S1核酸酶消化该环,即可进一步克隆。但自身引导合成法较难控制反应,而且用S1核酸酶切割发夹结构时无一例外地将导致对应于mRNA 5'端序列出现缺失和重排,因而该方法目前很少使用。 2、置换合成法 该方法利用第一链在反转录酶作用下产生的cDNA:mRNA杂交链不用碱变性,而是在dNTP存在下,利用RNA酶H在杂交链的mRNA链上造成切口和缺口。从而产生一系列RNA引物,使之成为合成第二链的引物,在大肠杆菌DNA聚合酶Ⅰ的作用下合成第二链。该反应有3个主要优点: (1) 非常有效; (2) 直接利用第一链反应产物,无须进一步处理和纯化; (3) 不必使用S1核酸酶来切割双链cDNA中的单链发夹环。 优化条件 模板 作为模板的RNA分子必须是完整的,并且不含DNA、蛋白和其他杂质。RNA中即使含有最微量的DNA,经扩增后也会出现非特异性扩增;蛋白若未除干净,与RNA结合后会影响逆转录和PCR;残留的RNase极易将模板RNA降解掉。 逆转录酶 1、目前商品化反转录酶有从禽类成髓细胞瘤病毒纯化到的禽类成髓细胞病毒(AMV)逆转录酶和从表达克隆化的Moloney鼠白血病病毒反转录酶基因的大肠杆菌中分离到的鼠白血病病毒(MLV)反转录酶。AMV反转录酶包括两个具有若干种酶活性的多肽亚基,这些活性包括依赖于RNA的DNA合成,依赖于DNA的 DNA合成以及对DNA:RNA杂交体的RNA部分进行内切降解(RNA酶H活性)。MLV反转录酶只有单个多肽亚基,兼备依赖于RNA和依赖于DNA的DNA合成活性,但降解RNADNA杂交体中的RNA的能力较弱,且对热的稳定性较AMV反转录酶差。 2、普通逆转录酶反应温度为37-42度,然而一些有丰富二级结构的复杂模板或者高GC含量的模板在常温下扩增困难,需要将逆转录反应置于较高温度下进行改善扩增。对于较高的反应温度,建议使用cMASTER-RT酶,该酶在37-60度的温度范围内都能保持良好的活性,使用cMASTER-RT酶,可以增加反应产量,还可以增加逆转录反应的特异性。因为,对于使用基因特异性引物(GSP)进行cDNA合成时,较高的反应温度允许使用Tm值较高的基因特异性引物。 Taq酶 普通的Taq酶扩增能力很强但是错配率高,虽然高产但是最大只能 扩增3-5kb的片段;而本身带有校读功能的高保真酶有很高的保真度,不易出错,可扩增较长的片段,但是产量偏低。TripleMaster酶则是一个高保真的混合酶—在保留Taq酶的高聚合能力,保证高产的同时,还有效降低了Taq的错配率,大大提高了保真性。 RT-PCR buffer 引物:(1)上下游引物设计在跨内含子的两个外显子的3’端和下一个外显子的5’端,这样不会在基因组上扩出来。(2)设计在两个离得远的外显子上,这样从基因组和cDNA上得到的片段长度不一样,可以一引两用。 Random 9mers 适用于长的及具有Hairpin构造的RNA.适用于rRNA、mRNA、tRNA等所有RNA的反转录反应 (用Random 9mers合成cDNA后,任何特异性的Primer pair都可用于PCR反应)。 Oligo dT-Adaptor Primer 适用于具有Poly(A)+ tail的RNA. 注: 原核生物的RNA、真核生物的rRNA及tRNA (某些种类真核生物的mRNA)不具有Poly(A) tail.本Primer设计巧妙,反转录效率高。反转录反应后,用M13 Primer M4可进行3'-RACE. 一步法和两步法 一步法: 利用同一缓冲液,在同一体系中加入逆转录酶、引物、Taq酶、4种dNTP 直接进行mRNA反转录与PCR扩增。发现Taq酶不仅具有DNA多聚酶的作用,而且具有反转录酶活性,可利用其双重作用在同一体系中直接以mRNA为模板进行反转录和其后PCR扩增,从而使mRNA的PCR步骤更为简化。所需样品量减少到最低限度,临床小样品的检测非常有利。用一步法扩增可检测出总RNA中小于1ng的低丰度mRNA.该法可用于低丰度mRNA的cDNA文库的构建及特异cDNA的克隆,并有可能与Taq酶的测序技术相组合,使得自动反转录、基因扩增与基因转录产物的测序在单管中进行。 两步法:由于单管反应时RT和PCR都不能在最佳条件下进行并且 容易相互干扰,常只适宜用基因特异引物扩增较短的基因,及定量PCR.两步法则是将RT和PCR分别进行,这样使得两个反应充分发挥各自的特点,更为灵活而且严谨,适合那些GC含量、二级结构严重的模板或者是未知模板,以及多个基因的RT-PCR。 |
|
兼并引物PCR(Degenerate Primer) 兼并引物是指代表编码单个氨基酸所有不同碱基可能性的不同序列的混合物。兼并度越低,产物特异性越强。设计引物时应尽量选择兼并性小的氨基酸,并避免引物3’末端兼并,因为3'端最后3个碱基的退火足以在错误位点起始PCR;次黄嘌呤可以同所有的碱基配对,降低引物的退火温度。 巢氏PCR(Nested PCR) 巢氏PCR需要两到三对引物,一般采用第一套引物扩增15-30个循环,再用扩增DNA片段内设定的第二套引物扩增15-30个循环,这样可使待扩增序列得到高效扩增,而次级结构却很少扩增。套式引物PCR减少了引物非特异性退火,从而增加了特异性扩增,提高了扩增效率。 若将套失PCR的内外引物稍加改变,延长外引物长度(25-30bp),同时缩短内引物长度(15-17bp),使外引物先在高退火温度下复性,做双温扩增,然后改换至三温循环,使内引物在外引物扩增的基础上,在低退火温度复性,直到扩增完成,这样就可以使两套引物一次同时加入。 反向PCR(Inverse PCR) 反向PCR的目的在于扩增一段已知序列旁侧的DNA,也就是说这一反应体系不是在一对引物之间而是在引物外侧合成DNA.反向PCR可用于研究与已知DNA区段相连接的未知染色体序列,因此又称为染色体步移。这时选择的引物虽然与核心DNA两末端序列互补,但引物3’端是相互反向的。 反向PCR的操作流程:扩增前先用限制性内切酶酶切样品DNA,然后用DNA连接酶连接成一个环状分子,通过反向PCR扩增引物的上游片段和下游片段。 选择多种限制性内切酶,基本准则是选择不能在非酶切位点切断靶DNA的酶。裂解核心区的内切酶使反向PCR只能扩增引物所定模板(依赖于引物)的上游或下游区,而不裂解核心区的酶则使两侧序列都扩增 Southern杂交来确定内切酶用以产生大小适于环化及反向PCR的片段的末端片段 大多数有核基因组含有大量中度和高度重复序列,所以往往需要两组到三组嵌套引物 不对称PCR(Asymmetric PCR) 不对称PCR的基本原理是采用不等量的引物产生大量的单链DNA(ss-DNA)。这两种引物分别称为限制性引物与非限制性引物;其最佳比例一般为1:50~1:100,关键是限制引物的绝对量。限制性引物太多太少,均不利于ss-DNA. 不对称PCR反应过程:一般来说,在不对称PCR反应中,在 开始的20-25个循环中,两个比率不对称的扩增引物产生出双链DNA,当限量的那个引 物耗光后,随后的5-10个循环就产生出ssDNA. 产生的ds-DNA与ss-DNA由于分子量不同,可以通过电泳分离。 ssDNA产物量 一般对100μl PCR反应体系而言,当引物比率为50pmo:0.5pmol,经过30个循环后,大约可产生1-3pmol的ssDNA.ssDNA的产量可用下列几种方法来估计: 1、在ss-DNA合成过程中,掺入32P-dNTP.取10%的反应产物,经过琼脂 糖电泳分离后,放射自显影。 2、取5%的反应物来走 胶,转移到膜上,并用与ssDNA互补的寡核苷酸为探针杂交。 解决不对称PCR反应扩增效率低的方法 增加PCR循环的次数 在最后五个循环中多加Tab聚合酶(2U) 试一下相反的不对称引物比率。有时,相反的不对称引物比率可能给出不同产量的ssDNA. 原位PCR 原位PCR就是在组织细胞里进行PCR反应,它结合了具有细胞定位能力的原位杂交和高度特异敏感的PCR技术的优点,是细胞学科研与临床诊断领域里的一项有较大潜力的新技术。 原位PCR是Hasse等于1990年建立的,实验用的标本是新鲜组织、石蜡包埋组织、脱落细胞、血细胞等。其基本方法为: 1、固定组织或细胞:将组织细胞固定于预先用四氟乙烯包被的玻片上,并用多聚甲醛处理,再灭活除去细胞内源性过氧化物酶。 2、蛋白酶K消化处理:用60ug/ml的蛋白酶K将固定好的组织细胞片55℃消化处理2h后,96℃2min以灭活蛋白酶K。 3、PCR扩增:在组织细胞片上,加PCR反应液,覆盖并加液体石蜡后,直接放在扩增仪的金属板上,进行PCR循环扩增。有的基因扩增仪带有专门用于原位PCR的装置。 4、杂交:PCR扩增结束后,用标记的寡核苷酸探针进行原位杂交。 5、显微镜观察结果。 原位PCR既能分辩鉴定带有靶序列的细胞,又能标出靶序列在细胞内的位置,于分子和细胞水平上研究疾病的发病机理和临床过程及病理的转归有重大的实用价值。其特异性和敏感性高于一般的PCR。
|
|
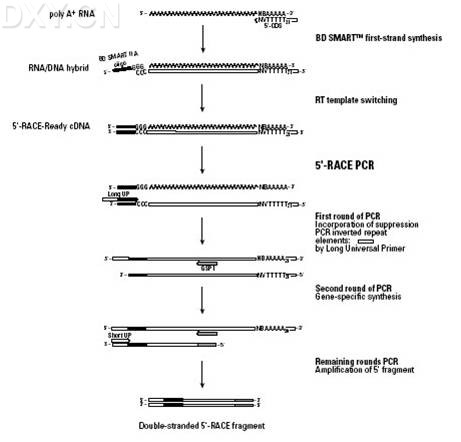
连接酶链反应(Ligase chain reaction,LCR) 连接酶链反应(Ligase chain reaction,LCR),是一种新的DNA体外扩增和检测技术,主要用于点突变的研究及靶基因的扩增。 连接酶链反应是Backman1997年为检出靶基因序列中的点突变而设计发明,并申报了专利。1988年Landegren也进行了该项研究。1988年Backman等又因分离热稳定的连接酶,而申报专利,1991年Backman和Barany分别用耐热DNA连接酶进行了LCR试验。耐热DNA连接酶可以在热循环中保持活性,提高连接反应的特异性,排除了背景扩增和免除了不断补充酶的繁琐程序。 LCR的基本原理为利用DNA连接酶。特异地将双链DNA片段连接,经变性-退火-连接三步骤反复循环,从而使靶基因序列大量扩增。其程序为:在模DNA、DNA连接酶、寡核苷酸引物以及相应的反应条件下,首先加热至一定温度下(94~95℃)使DNA变性,双链打开,然后降温退火(65℃),引物与之互补的模板DNA结合并留下一缺口,如果与靶序列杂交的相邻的寡核苷酸引物与靶序列完全互补,DNA连接酶即可连接封闭这一缺口,则LCR反应的三步骤(变性-退火-连接)就能反复进行,每次连接反应的产物又可在下一轮反应中作模板,使更多的寡核苷酸被连接与扩增。若连接处的靶序列有点突变,引物不能与靶序列精确结合,缺口附近核苷酸的空间结构发生变化,连接反应不能进行,也就不能形成连接产物。 LCR的引物是两对分别互补的引物,引物长度为20~26个,以保证引物与靶序列的特异性结合,LCR识别点突变的特异性高于PCR,其特异性首先取决于引物与模板的特异性结合,其次是耐热连接酶的特异性。LCR连接反应温度接近寡苷酸的解链温度(Tm),因而识别单核苷酸错配的特异性极高。 LCR的扩增效率与PCR相当,用耐热连接酶做LCR只用两个温度循环,94℃min变性和65℃复性并连接,循环30次左右。其产物的检测也较方便灵敏。目前该方法主要用点突变的研究与检测、微生物病原体的检测及定向诱变等,还可用于单碱基遗传病多态性及单碱基遗传病的产物诊断,微生物的种型鉴定,癌基因的点突变研究等。 cDNA末端快速扩增技术(rapid amplification of cDNA ends, RACE) cDNA末端快速扩增技术(rapid amplification of cDNA ends, RACE)是一种基于PCR从低丰度的转录本中快速扩增cDNA的5'和3’末端的有效方法 5’ RACE-PCR 利用mRNA的3‘末端的poly(A)尾巴作为一个引物结合位点,以Oligo(dT)30MN作为锁定引物在反转录酶MMLV作用下,反转录合成标准第一链cDNA.利用该反转录酶具有的末端转移酶活性,在反转录达到第一链的5‘末端时自动加上3一5个(dC)残基,退火后(dC)残基与含有SMART寡核苷酸序列Oliogo(dG)通用接头引物配对后,转换为以SMART序列为模板继续延伸而连上通用接头(figure 2 )。然后用一个含有部分接头序列的通用引物UPM(universal primer,UPM)作为上游引物,用一个基因特异引物2(GSP 2 genespecific primer,GSP)作为下游引物,以SMART第一链cDNA为模板,进行PCR循环,把目的基因5‘末端的cDNA片段扩增出来。最终,从2个有相互重叠序列的3' / 5‘-RACE产物中获得全长cDNA,或者通过分析RACE产物的3‘和5‘端序列,合成相应引物扩增出全长cDNA.
3’ RACE-PCR 利用mRNA的3‘末端的poly(A)尾巴作为一个引物结合位点,以连有SMART寡核营酸序列通用接头引物的Oligo(dT)30MN作为锁定引物反转录合成标准第一链cDNA.然后用一个基因特异引物GSP1(gene specific primer,GSP)作为上游引物,用一个含有部分接头序列的通用引物UPM(universal primer,UPM)作为下游引物,以cDNA第一链为模板,进行PCR循环,把目的基因3‘末端的。DNA片段扩增出来。
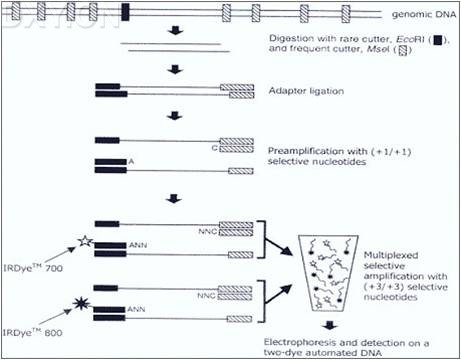
AFLP(扩增性片段长度多态性 ) AFLP技术是一项新的分子标记技术,是基于PCR技术扩增基因组DNA限制性片段,基因组DNA先用限制性内切酶切割,然后将双链接头连接到DNA片段的末端,接头序列和相邻的限制性位点序列,作为引物结合位点。限制性片段用二种酶切割产生,一种是罕见切割酶,一种是常用切割酶。它结合了RFLP和PCR技术特点,具有RFLP技术的可靠性和PCR技术的高效性。由于AFLP扩增可使某一品种出现特定的DNA谱带,而在另一品种中可能无此谱带产生,因此,这种通过引物诱导及DNA扩增后得到的DNA多态性可做为一种分子标记。 AFLP可在一次单个反应中检测到大量的片段。以说AFLP技术是一种新的而且有很大功能的DNA指纹技术。
免疫-PCR(immuno-PCR) 免疫-PCR(immuno-PCR)是新近建立的一种灵敏、特异的抗原检测系统。它利用抗原-抗体反应的特异性和PCR扩增反应的极高灵敏性来检测抗原,尤其适用于极微量抗原的检测。 免疫-PCR试验的主要步骤有三个:①抗原-抗体反应,②与嵌合连接分子结合,③PCR扩增嵌合连接分子中的DNA(一般为质粒DNA)。该技术的关键环节是嵌合连接分子的制备。在免疫-PCR中,嵌合连接分子起着桥梁作用,它有两个结合位点,一个与抗原抗体复合物中的抗体结合,一个与质粒DNA结合,其基本原理与ELISA和免疫酶染色相似,不同之处在于其中的标记物不是酶而是质粒DNA,在操作反应中形成抗原抗体-连接分子-DNA复合物,通过PCR扩增DNA来判断是否存在特异性抗原。 免疫PCR优点为:①特异性较强,因为它建立在抗原抗体特异性反应的基础上。②敏感度高,PCR具有惊人的扩增能力,免疫PCR比ELISA敏感度高105倍以上,可用于单个抗原的检测。③操作简便,PCR扩增质粒DNA比扩增靶基因容易得多,一般实验室均能进行。 MSP甲基化特异PCR 一般调控区保持甲基化状态,基因不表达 直接干扰转录因子和启动子识别位点的结合 与转录抑制因子结合引起基因沉默 改变染色质的结构 MSP甲基化特异PCR是用对苯二酚和亚硫酸氢钠处理氢氧化钠变性的基因组DNA,使得未甲基化的C转化为T.按照C转换T后的基因序列设计出来的引物扩出目的基因。由于甲基化CpG岛中的C不能被转化为T,用以上的引物将不会扩出目的基因。通过上述手段就能达到识别基因组DNA甲基化与否的目的。 |
→如果您认为本词条还有待完善,请 编辑词条
词条内容仅供参考,如果您需要解决具体问题
(尤其在法律、医学等领域),建议您咨询相关领域专业人士。
1
收藏到: