|
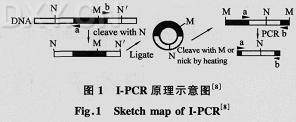
随着基因工程的发展,常常需要构建一种能高水平表达异源蛋白质的表达载体。启动子对外源基因的表达水平影响很大,是基因工程表达载体的重要元件。因此研究启动子的克隆方法,对研究基因表达调控和构建表达载体至关重要。 迄今为止,国外尚未见到有关启动子克隆方法的综述性报道,国内仅孙晓红等曾就启动子的结构、分类、克隆方法和食用菌中已经分离到的启动子作过综述。而近年来又有许多改进的克隆启动子的方法获得了多方面的成功,本文就近年来改进的启动子克隆方法作一综述,以期促进对启动子分离技术的应用。 1 启动子克隆的几种方法 1.1 利用启动子探针载体筛选启动子 启动子探针型载体是一种有效、经济、快速分离基因启动子的工具型载体,包含2个基本部分:转化单元和检测单元。其中,转化单元含复制起点和抗生素抗性基因,用于选择被转化的细胞;检测单元则包括1个已失去转录功能且易于检测的遗传标记基因以及克隆位点。 利用启动子探针载体筛选启动子的过程为,先选用1种适当的限制性核酸内切酶消化切割染色体DNA,然后将切割产生的DNA限制片段群体与无启动子的探针质粒载体重组,并按照设计的要求使克隆的片段恰好插在紧邻报告基因的上游位置;随后再把重组混合物转化给寄主细胞,构建质粒载体基因文库,并检测报告基因的表达活性。 当插入段同时满足(1)具有基因启动子序列;(2)具有翻译启始区;(3)具有启始密码子;(4)插入方向正确;(5)插入片段3'端编码区序列抗性基因编码区读码框一致,则有可能形成有功能的抗性融合基因,从而启动抗性基因的表达。 最早由Rachael等在大肠杆菌中以四环素抗性基因作为报告基因构建了启动子探针质粒pBRH3B,并克隆了一些原核和真核启动子片段。其后Donna等以氯霉素抗性基因作为报告基因,Fodor等以大肠杆菌LacZ为报告基因,构建了酵母启动子探针质粒并克隆了一些启动子片段。构建启动子探针型载体,较为常见的检测标记基因有β-半乳糖苷酶基因(lacZ)、氯霉素乙酰转移酶基因(cat)、四环素抗性基因(Tet')和卡那霉素抗性基因(Kan')。近年来,人们渐渐较多地使用潮霉素B磷酸转移酶(hph)基因作为检测标记基因。李维等曾构建了含有hph抗性基因的启动子探针型载体pSUPV8,直接在大肠杆菌中分离黄孢原毛平革菌基因的启动子。先用Sau3AI酶切黄孢原毛平革菌基因总DNA,再与用BamHI酶切后的pSUPV8相连,转化大肠杆菌,用间接筛选法从氨苄青霉素和潮霉素抗性平板上筛选重组子,得到6个双抗重组子(pCH1~pCH6),电泳检测插入片段分别命名为CHl~CH6;再用原生质体转化法将重组子分别转化黄孢原毛平革菌,对获得的转化子进行复筛,仅pCH6的转化平板上有稳定生长的菌落,说明了CH6片段在黄孢原毛平革菌中具有启动基因表达的功能。该方法不需要知道具体基因的序列,可随机筛选启动子,避免了引物设计,能获得大量的启动子片段。 1.2 利用PCR技术克隆启动子 即根据发表的基因序列,设计引物,克隆基因的启动子,由于PCR法简便快捷,近年来人们较多采用此方法克隆基因启动子。 苏宁等根据已报道的水稻叶绿体16SrRNA启动子基因序列设计5'启动子序列的引物,以水稻叶绿体DNA为模板,PCR扩增出16SrRNA基因5'启动子区的片段,酶切克隆到pSK的SacI和SphI位点,构建测序载体质粒pZ16S,进行序列测定,结果表明所克隆的片段长为144bp,含有SD序列。同源比较结果表明,所克隆的片段与水稻叶绿体16SrRNA启动子序列具有100%的同源性。 上述的PCR方法简便、快捷、操作简单,是人们较为广泛使用的技术。 1.3 环状 PCR 环状PCR包括I-PCR(Inverse-PCR)和P-PCR(Panhandle-PCR)。这2种PCR都是根据一端已知序列设计的嵌套式引物进行PCR。 1.3.1 I-PCR I-PCR是1988年由Triglia最早提出的一种基于PCR的改进的染色体步行方法。 I-PCR的实验程序包括,基因组DNA经酶切后用T4DNA连接酶进行自连接,产生环状DNA片段;以环化产物为底物,用根据已知片段设计的反向引物进行PCR扩增,从而得到含有未知片段的扩增产物(流程如图1所示)。
|
|
韩志勇等以I-PCR技术为基础克隆了转基因水稻的外源基因旁侧序列。先用小量法提取转基因水稻的总DNA,总DNA用10倍过量的限制内切酶进行过夜酶切,酶切片段进行自连接,然后根据工程质粒的T-DNA区设计2对反向引物,进行套式PCR扩增旁侧序列。建立了适合于处理大量材料的克隆转基因水稻中外源基因旁侧序列的技术体系。在1周内克隆了35个转基因水稻株系中外源基因的旁侧序列,长度在300~750bp之间。I-PCR法快速、高效、稳定,操作相对简单,花费少,PCR引物设计比较方便。 1.3.2 P-PCR P-PCR是由Jones等提出的利用末端反向重复序列与已知序列互补配对形成环状单链模板,有效增强了引物与模板结合的特异性。反应需要3个根据已知序列设计的引物,3个引物在已知序列内呈线性排列,其中第3个引物可作为接头使用,可与已知序列互补配对形成锅柄状单链模板。其过程为,首先酶切基因组DNA,产生5'或3'粘末端,然后连接上合适的接头(primer 3),连接好后最好用核酸外切酶I除去多余的接头,由于连接上的接头与已知序列是反向重复序列,变性后的DNA单链可退火形成锅柄状单链模板,之后分别用3个单引物进行3次PCR扩增,能有效地扩增2~9kbp的大片段未知序列(流程如图2所示)。
黄君健等成功地应用P-PCR技术从正常的人外周血单核细胞基因组DNA中扩增端粒催化亚基hTERT基因5'端上游旁侧序列,获得了hTERT基因翻译启始位点上游2090bp的基因组DNA序列。首先用酶切消化基因组DNA,得到带有GATC的5'突出端的DNA片段。然后利用已知的hTERTcDNA序列设计PCR引物,用常规的PCR方法扩增出1条大约900bp的基因组特异片段,序列分析为hTERT的基因组DNA片段。根据得到的基因组DNA序列的信息,确定P-PCR的引物退火区,并合成了5'磷酸化的连接寡核苷酸和4条基因特异性引物,其中连接寡核苷酸5'端的4个碱基CTAG与上述核酸内切酶消化产生的5'突出端GATC互补,然后将连接寡核苷酸与基因组酶切产物连接,以连接产物为反应模板,进行PCR,使模板自身进行退 火-延伸反应,以形成Panhandle结构。最后以单链Panhandle为模板,4条基因特异序列为引物进行嵌套式PCR,最终获得了1条约2kb的含hTERT基因启动子的DNA片段。Jones等利用改进的P-PCR,在形成panhandle结构之前3'末端连上ddCTP,使引物错配的机率减少,特异性增加。他们从人类基因组DNA已知位点侧翼扩增了4~9kb的大片段未知序列。P-PCR是目前能够扩增距已知序列最远的未知DNA序列的方法,有很高的特异性。 1.4 利用载体或接头的染色体步行技术克隆基因启动子 这类方法的第一步都是酶切基因组DNA,连接载体或接头,既可以用pUCl8等质粒载体,也可以使用λDNA等噬菌体载体,只要选用的载体带有合适的酶切位点;同样根据实验需要,接头既可以是双链也可以是单链,然后根据基因组DNA序列设计的特异引物和载体的通用引物或接头序列进行扩增。 1.4.1 利用载体的PCR Shyamala等利用的单特异性引物PCR(SSP-PCR)对以小鼠伤寒杆菌组氨酸转运操纵子为起点进行连续步行。以M13mpl8RF DNA为载体。用PstI和AraI酶切基因组DNA,PstI和XmaI酶切载体DNA,然后连接基因组片段和载体片段,用根据基因组DNA序列设计的特异引物和载体的通用引物进行扩增,由于非特异片段没有单特异引物结合的位点,即使有载体连到非特异片段,也无法得到大量扩增,而使特异片段得到有效扩增。 |
|
1.4.2 利用接头的PCR王新国等利用衔接头的方法,设计了位于单链DNA两端互补的颠倒末端重复序列,增加了反应的特异性,在胡萝卜II型转化酶基因启动子的克隆方面取得了新的进展。首先将胡萝卜基因组DNA分别用PvuI、SmaI、DraI、EcoRV酶切,并设计了1个衔接头长链序列和1个衔接头短链序列,并在衔接头短链的3'末端带有1个氨基的衔接头,能够阻止聚合酶催化的衔接头短链的延伸,同时衔接头的长链和短链之间是反向重复序列。将酶切片段与此衔接头连接,取连接产物做模板,以衔接头引物和基因特异引物做PCR,在首轮PCR中只有限定的远端基因特异引物有结合位点,当基因特异引物延伸产生的DNA链通过衔接头时,才能产生衔接头引物的结合位点,PCR才能以衔接头引物和基因特异引物进行指数扩增。而另一方面,如果非特异合成产生了DNA两端都有双链衔接头序列的PCR产物时,这种PCR产物在每次变性后,单链DNA末端的衔接头反向重复序列将形成锅柄结构,此结构比引物-模板杂交更稳定,能抑制非特异序列的指数增长。最后得到主要的PCR产物为3.4kb、1.3kb、0.6kb和0.4kb。将EcoR V-衔接头体系的PCR产物克隆、测序、同源性比较,得到1个新的胡萝卜II型转化酶基因启动子序列,它含有类似于TATA box和CAAT box的元件,在启动子的远上游区域含多个AT富含区,该启动子的发现对于研究植物中的糖代谢具有重要的意义。接头引物的相对位置如图3所示。
这种方法具有便于操作、实验线路简单的优点,但是特异性较差,产物需进一步杂交验证。 1.5 YADE法 Prashar等在扩增cDNA3'端时采用“Y”形接头,以减少接头引物的单引物扩增。 其原理是接头引物处于“Y”接头的2个分叉单链上,序列与接头一样,只有与特异引物引导合成了接头的互补序列后,接头引物才能退火参与扩增,流程如图4。
方卫国等尝试将YADE法引入到昆虫病原真菌的分子生物学研究,并取得了成功,建立了适合于球孢白僵菌和金龟子绿僵菌YADE体系。在已克隆的类球孢白僵菌类枯草杆菌蛋白酶基因CDEP-1的基础上,利用YADE法,克隆到该基因的启动子CDEPP。 先酶切球孢白僵菌基因组 DNA,然后与“Y”形接头相连,取连接产物做模板,先以基因特异引物1做线性扩增,再以线性扩增产物为模板,以接头引物和基因特异引物2做指数扩增,只有当线性扩增时合成了含有接头引物的互补单链,接头引物才能与其发生退火,参与指数扩增,从而有效防止了接头引物的单引物扩增。最后得PCR产物,进行序列分析确定为CDEP-1的上游启动子序列。 在应用YADE法时,内切酶的选择至关重要。好的内切酶产生适合PCR扩增的片段,太大太小都不行。为了得到合适的内切酶,需要从众多的内切酶中筛选。研究表明,不同的物种有自己合适的内切酶。YADE法延伸的起始片段可以是基因组 DNA片段,也可以是cDNA片段,在延伸cDNA片段时,设计的引物需要避开内含子和外显子的边界,在内含子的位置未知的情况下,可考虑多合成1~2条特异引物,以提高扩增未知片段的机率。该方法假阳性低、效率高,理论上能扩出所有目的片段。 |
|
1.6 TAlL-PCR 很早就有用随机引物的PCR,但由于无法有效地控制由随机引物引发的非特异产物的产生,所以一直未能广泛应用。近年来由IJiu等设计的TAIL-PCR(Termal Asymmetric Interlaced PCR)又叫热不对称交错PCR,则解决了这个问题,后来有研究表明,经改良过的TAIL-PCR成功地从突变体中克隆到外源插入基因的旁侧序列,从而为启动子的克隆提供了有效的新方法。 在利用特异引物和随机引物进行PCR中一般有3种产物生成:(1)由特异性引物和简并引物扩增出的产物;(2)由同一特异性弓l物扩增出的产物;(3)由同一简并引物扩增出的产物。在TAIL-PCR反应中,其中后2种目标产物可以通过以嵌套的特异性引物进行的后续反应来消除。 TAIL-PCR的基本原理是利用目标序列旁的已知序列设计3个嵌套的特异性引物(specialprimer,简称sp1,sp2,sp3,约20bp),用它们分别和1个具有低Tm值的短的随机简并引物(Arbitrarydegenerate prime,AD,约14bp)相组合,以基因组DNA为模板。根据引物的长短和特异性的差异设计不对称的温度循环,通过分级反应来扩增特异引物(流程如图5所示)。
TAIL-PCR共分3次反应。第一次反应包括5次高特异性、1次低特异、10次较低特异性反应和12个热不对称的超级循环。5次高特异性反应,使sp1与已知的序列退火并延伸,增加了目标序列的浓度;1次低特异性的反应使简并引物结合到较多的目标序列上;10次较低特异性反应使2种引物均与模板退火,随后进行12次超级循环。 经上述反应得到了不同浓度的3种类型产物:特异性产物(I)型和非特异性产物(Ⅰ型和Ⅲ型)。第二次反应则将第一级反应的产物稀释1000倍作为模板,通过10次热不对称的超级循环,使特异性产物被选择地扩增,而非特异产物含量极低。第三次反应又将第二次反应的产物稀释作模板,再设置普通的PCR反应或热不对称超级循环,通过上述3次PCR反应可获得与已知序列邻近的目标序列。 |
|
Gento等曾用构建的含有潮霉素抗性基因(hph)的双元表达载体pBIG2RHPH2转化真菌,然后利用TAIL-PCR法克隆得到的真菌转化子基因组DNA的T-DNA插入区的旁侧序列并取得了成功。根据T-DNA区的HPH基因设计了扩增右边界的3个引物HS1~HS3,以及扩增左边界的引物HAS2~HAS4,另外又根据不同的转化子分别设计了简并引物ADl~AD3(引物位置如图6所示)。
在首轮PCR中,以AD/HS1为引物扩增右边界(以AD/HAS2扩增左边界),然后取首轮PCR产物为模板,以AD/HS2(AD/HAS3)进行二次PCR,再以二次PCR产物为模板,AD/HS3(AD/HAS4)为模板进行第三轮。 PCR,将3轮的PCR产物进行电泳分析结果表明,采用TAIL-PCR的方法成功地从突变体中获得了带有T-DNA左右边界的旁侧序列,从而证明了TAIL-PCR法是有效地扩增基因旁侧序列的方法,为启动子的克隆又增添了1种可行的方法。 TAIL-PCR不需要PCR前的任何DNA操作,避免了环化和连接,速度快,特异性强,效率高,灵敏,在分子生物学研究的各个领域都有广泛的应用。 2 讨论 以上介绍的几种方法基本代表了现有的启动子克隆方法,它们分别具有不同的特点和适用范围。 利用启动子探针载体筛选启动子时,不需要知道具体的基因序列,避免了引物设计,并能获得大量的启动子片段;其缺点是需要构建1个穿梭质粒,建库、转化、筛选,工作量大,费时费力,而且克隆、亚克隆的过程繁琐。因此在基因的遗传背景不是很清楚时,往往通过探针载体随机筛选启动子。 而PCR法的主要优点是简便、快捷、操作简单;其缺点是只能扩增两端已知序列间的DNA区,且扩增的特异性较低。其适用条件是建立在对基因序列十分清楚的基础上,只有知道基因的全序列,才可根据已知序列设计引物,扩增出该基因的启动子。因此,在基因序列清楚的情况下.我们首先想到的就是PCR法。 I-PCR法操作简单,克服了文库筛选、克隆、亚克隆的繁琐步骤,对实验条件要求不高,花费少,在PCR法的基础上,增加了DNA的环化过程,从而可以扩增只有一端序列已知的DNA,由于不需要设计和合成大量昂贵的核苷酸接头,因此避免了引物设计和有接头而产生的非特异性产物的麻烦;然而由于直接克隆已知序列外的未知DNA区域依赖于DNA的环化性,而环化连接过程中常常产生多联体,成为副产物甚至是主要产物,导致非特异扩增,造成了闭环双链的DNA的PCR扩增效率差,经常得不到满意的结果,同时酶切片段太长也会使扩增效率下降。其适用于寻找只有一端序列已知的DNA,对未知DNA片段进行扩增。 相对于I-PCR,P-PCR经过设计形成的是锅柄状单链DNA模板,从而有效增加了引物与模板结合的特异性,p-PCR能够完成全部嵌套式扩增,因而产物有非常高的特异性;其缺点是也存在DNA环化、连接的弊端,但与I-PCR相比,其PCR产物的特异性已大大增加。目前,P-PCR可以扩增位于已知位点侧翼的大于3.0kb的人基因组DNA的启动子,是目前为止能扩增距离已知序列最远的DNA序列的方法.因而常用于扩增大片段的未知序列。 利用载体的PCR克隆启动子有实验设计简便的优点,在基因组DNA中加入含合适酶切位点的载体,是基于PCR法的又一创新之处;然而其实验操作较为繁琐,特异性差,即使用套式PCR,仍然有几条电泳条带,因而特异性产物需杂交进一步确定。 利用接头的PCR克隆启动子的优点是避免了DNA环化,改进的接头可以通过形成锅柄结构有效抑制非特异性序列的扩增,但设计和合成大量的核苷酸接头,价格昂贵;此外用常规的加接头的方法来克隆。 |
|
启动子时往往有接头非特异产物产生,特异性较低,特异产物需杂交确定。由于利用接头的PCR法不需要DNA环化,通常适用于寻找已知cDNA周围未知启动子或其它调控区域。 YADE法在利用接头PCR时,巧妙地设计了“Y”型接头,从而有效地防止接头引物的单引物扩增,延伸时起始片段可以是基因组DNA也可以是cDNA.可用于复杂的基因组的PCR步行,能广泛应用于真核生物;其缺点是成本较高,在应用YADE法时,为了得到合适的内切酶,需要从众多的内切酶中筛选,另外,特殊的接头往往也增加了实验的成本。通常对于较复杂的真核生物基因组可以采用YADE法。 目前理想的启动子克隆方法需要更为广泛的,不需要PCR前的酶切、环化等操作,且特异性较高的PCR技术,TAIL-PCR基本符合这一条件。TAIL-PCR法的有以下优点:(1)方法简单,只要设计好引物,即可用基因组DNA为模板直接进行PCR扩增;(2)特异性高,用简并引物和特异性嵌套引物相组合,通过不对称的温度循环和分级反应,使最终的目的片段占绝对优势;(3)高效灵敏,使用任何一个AD引物,在60%~80%的反应中能产生特异性产物;(4)快速,整个反应可在ld内完成,(5)避免了DNA的环化和连接,反应产物准确可靠,重复性好。TAIL~PCR的创新之处在于其热不对称的分级反应,有效防止了非特异产物的扩增;但是TAIL~PCR仍有很多不尽人意的地方:TAIL~PCR反应需要较多的引物组合;此外,由于随机简并引物存在有限的结合位点,对个别侧翼序列,即使使用不同的简并引物也难以扩增到阳性结果,整个反应需要一系列连续反应,条件设置要求精细;还要求引物和模板有较高的纯度,如果有降解或纯度不够,也很难扩增出特异性条带;此外,TAlL~PCR还很有可能扩增的是高度重复序列,因而无法步行。TAIL~PCR技术适合于分离获得克隆载体上的DNA序列,达到克隆相关基因的启动子的目的,也可用于基因组小的物种,如拟南芥和基因组大的物种,如小麦的已知序列两侧翼的DNA序列的分离。 3 展望 目前,启动子克隆的方法种类繁多,进展迅速。启动子克隆方法绝大多数是建立在PCR的基础上的染色体步行技术,因而利用启动子克隆的方法不仅可以克隆到基因的启动子,同时还可以用于克隆cDNA的全长。分子生物学的一个重要问题是如何利用大量积累的基因部分序列(如EST)进一步克隆全长基因及其调控序列,而启动子克隆方法为这一问题提供了简便有效的手段。另外,随着基因工程的发展,出现了越来越多的转基因动植物,启动子克隆的方法也可以应用在转基因生物的研究中,如转基因水稻的T-DNA区,通过启动子克隆方法的PCR步行技术,能成功地克隆到T-DNA区的旁侧序列,有助于分析突变体中突变区序列。 随着PCR技术的不断进步,一定会有更多、更好的克隆启动子的方法产生,而它们最后也将会是克隆基因全长或突变体(特别是转座子插入突变体)中目的基因的简便高效的新方法。 注: (1)参考文献:略;需者可与本站Email联系,或到中国水稻研究所图书馆查阅; (2)文章来源:中国生物工程杂志,2005年25卷第7期; (3)作者单位:黑龙江大学生命科学学院等。 |
→如果您认为本词条还有待完善,请 编辑词条
上一篇DNA重组实验常见问题分析 下一篇基因诊断技术的选择
词条内容仅供参考,如果您需要解决具体问题
(尤其在法律、医学等领域),建议您咨询相关领域专业人士。
0
收藏到: