|
INTRODUCTION After chromatin immunoprecipitation (ChIP), different PCR-based approaches can be used to determine how much DNA is precipitated at a locus of interest. Real-time PCR amplification is often the preferred technique. One can also use duplex PCR amplification, which is the coamplification of a fragment from the region of interest and a control fragment (e.g., the actin gene, or the tubulin gene). This approach allows for estimating relative levels of specific histone modifications along chromosomal domains. For allele-specific studies (for instance, on dosage-compensation mechanisms or on genomic imprinting), electrophoretic detection of single-strand conformation polymorphisms (SSCP) or similar strategies such as hot-stop PCR can differentiate PCR products that represent the silent allele from those amplified from the active allele. If a polymorphic restriction site is present in one allele and absent in the other, the method of choice is hot-stop PCR. If no polymorphic restriction sites are available, but there are single nucleotide polymorphisms (SNPs) that distinguish the alleles of interest, the best approach is to separate the PCR products derived from the two different alleles using SSCP. In SSCP, it is possible to discriminate denatured PCR products derived from one allele or the other because the secondary structure of each single strand will be directly dependent on the sequence itself. Hence, in nondenaturing gel conditions, each single strand will migrate differently. These four PCR-based methodologies to analyze immunoprecipitated chromatin (real-time PCR, duplex PCR, hot-stop PCR, and SSCP) are presented here. RELATED INFORMATION Our method for chromatin immunoprecipitation (ChIP) is described in Chromatin Immunoprecipitation on Unfixed Chromatin from Cells and Tissues to Analyze Histone Modifications. To distinguish between alleles at loci of interest in precipitated chromatin fractions, we use "hot-stop" PCR (Steps 1-22) or SSCP (Steps 23-37). See Uejima et al. (2000) for a detailed hot-stop PCR protocol. Figure 1 provides an example of results using SSCP. To quantify how much DNA has precipitated at a locus of interest, we use real-time PCR (Steps 38-39) and duplex PCR (Steps 40-53). Examples of duplex PCR analysis of precipitated chromatin are provided in Noma et al. (2001) and Gregory et al. (2001).
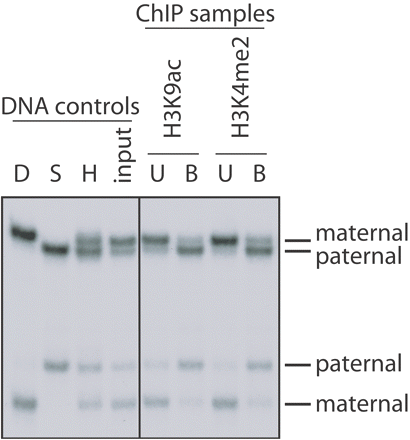
Figure 1. Allele-specific patterns of histone modifications revealed by PCR amplification and SSCP electrophoresis. Lung tissue was dissected from a mouse that was an interspecific hybrid (H) between Mus musculus domesticus (D, paternal genome) and Mus spretus (S, maternal genome). The native chromatin was then immunoprecipitated with rabbit polyclonal antisera to acetylation at lysine 9 of H3 (H3K9Ac) and to dimethylation at lysine 4 of H3 (H3K4Me2; Upstate Ltd.). Radioactive PCR was performed on bound (B) and unbound (U) fractions with primers that amplified from a unique sequence at an imprinting-control center located in a gene called Kvlqt1. The PCR products were denatured and subjected to electrophoresis through a nondenaturing polyacrylamide gel (SSCP electrophoresis). The four lanes to the left show control amplifications from genomic DNAs (D, Mus musculus domesticus DNA; S, Mus spretus DNA; H, [Mus musculus domesticus x Mus spretus] F1 DNA). In the analysis of the antibody bound (B) and unbound (U) fractions (right panel), the bands representing the maternal and paternal alleles are indicated. MATERIALS Reagents 100-bp DNA step ladder (for duplex PCR only; see Steps 40-53) Acrylamide solution for SSCP gels (2X) (e.g., Acrylamide Solution for Mutation Detection, A5934, Sigma) (for SSCP only; see Steps 23-37) MDE Gel Solution is a polyacrylamide-like matrix specifically optimized for SSCP. Acrylamide/bisacrylamide (29:1 ratio; 40% stock solution) (Sigma) (for hot-stop PCR only; see Steps 1-22) Agarose (for duplex PCR only; see Steps 40-53) Ammonium persulfate (APS; 10%, w/v), freshly prepared (for hot-stop PCR [Steps 1-22] or SSCP [Steps 23-37] only) α-32PdCTP (10 µCi/µl, specific activity 3000 Ci/mmol) (for hot-stop PCR [Steps 1-22] or SSCP [Steps 23-37] only) dNTPs (stock solutions at 25 mM for each dNTP) DNA loading buffer (6X) 30% (v/v) glycerol 0.25% (w/v) bromophenol blue 0.25% (w/v) xylene cyanol FF Store at 4°C Ethidium bromide solution (20 mg/ml in H2O) (for duplex PCR only; see Steps 40-53) Forward and reverse primers (100 µM stock solutions in H2O) For duplex PCR (Steps 40-53), primers should be designed in order to obtain comparable amplifications of the specific fragment of interest and control fragments (e.g., the actin gene) when using a control genomic DNA as a template. Importantly, the PCR product amplified from the region of interest should be of a size different from that amplified from the internal control region. This allows the two different PCR products to be distinguished by agarose gel electrophoresis. PCR amplification buffer (10X) (supplied with the Taq DNA polymerase) Reagents for Real-Time PCR (for real-time PCR only; see Steps 38-39) Restriction endonuclease (for hot-stop PCR only; see Steps 1-22) This endonuclease must be specific for a polymorphic restriction site within the amplified DNA fragment. Restriction endonuclease buffer (10X) (supplied with the restriction endonuclease; for hot-stop PCR only; see Steps 1-22) SSCP loading dye (for SSCP only; see Steps 23-37) 95% (v/v) formamide 10 mM NaOH 0.25% (w/v) bromophenol blue 0.25% (w/v) xylene cyanol Taq DNA polymerase (5 U/µl) TBE Buffer (1X and 5X) Prepare a 5x stock solution in 1 liter of H2O: 54 g of Tris base 27.5 g of boric acid 20 ml of 0.5 M EDTA (pH 8.0) The 0.5x working solution is 45 mM Tris-borate/1 mM EDTA. TBE is usually made and stored as a 5x or 10x stock solution. The pH of the concentrated stock buffer should be approx. 8.3. Dilute the concentrated stock buffer just before use and make the gel solution and the electrophoresis buffer from the same concentrated stock solution. Some investigators prefer to use more concentrated stock solutions of TBE (10x as opposed to 5x). However, 5x stock solution is more stable because the solutes do not precipitate during storage. Passing the 5x or 10x buffer stocks through a 0.22-µm filter can prevent or delay formation of precipitates. Template DNA This is the genomic DNA extracted from the antibody-bound and antibody-unbound fractions (from Chromatin Immunoprecipitation on Unfixed Chromatin from Cells and Tissues to Analyze Histone Modifications). Control genomic DNA should be used as well. For each PCR reaction, we use 20-50 ng of template DNA. N,N,N',N'-Tetramethyl-ethylenediamine (TEMED) (for hot-stop PCR [Steps 1-22] or SSCP [Steps 23-37] only) |
|
Equipment Cassettes with scintillation screens for exposure of X-ray films (for hot-stop PCR [Steps 1-22] or SSCP [Steps 23-37] only) Gel dryer for acrylamide gels (for hot-stop PCR [Steps 1-22] or SSCP [Steps 23-37] only) Imaging equipment for densitometric measurements on exposed X-ray films (for hot-stop PCR [Steps 1-22] or SSCP [Steps 23-37] only) Imaging equipment for agarose gels, and computer software for analyzing band intensities (for duplex PCR only; see Steps 40-53) PCR tubes (0.2-ml thin-walled) PhosphorImager (optional; see Steps 21 and 37) Horizontal gel electrophoresis tank for agarose gels (for duplex PCR only; see Steps 40-53) Standard DNA sequencing gel apparatus with 31 x 38.5-cm glass plates, 0.4-mm spacers, and a shark's-tooth comb (for hot-stop PCR [Steps 1-22] or SSCP [Steps 23-37] only) Thermal cycler Thin transparent plastic wrap (e.g., Saran Wrap) (for hot-stop PCR [Steps 1-22] or SSCP [Steps 23-37] only) Vertical gel electrophoresis tank for polyacrylamide gels with 21.7 x 16.5-cm glass plates, 0.4-mm spacers, a shark's-tooth comb, and clamps (for hot-stop PCR [Steps 1-22] or SSCP [Steps 23-37] only) Water bath (variable temperature) Whatman 3MM paper (for hot-stop PCR [Steps 1-22] or SSCP [Steps 23-37] only) X-ray film (for hot-stop PCR [Steps 1-22] or SSCP [Steps 23-37] only) METHOD Analysis of precipitated chromatin fractions (from Chromatin Immunoprecipitation on Unfixed Chromatin from Cells and Tissues to Analyze Histone Modifications) by PCR requires extreme care. Depending on the amount of input chromatin and the abundance of the modification at the site of interest, sometimes only a small amount of DNA template will be available for amplification. We recommend taking all possible precautions to prevent contamination from other DNA sources: amplification in a dedicated space (PCR hood), use of a set of dedicated pipettes for setting up PCR reactions only, use of filtered pipette tips, and so on. Allele-Specific PCR Analysis of Precipitated Chromatin Hot-Stop PCR Amplification Place 50-100 ng of template DNA in a 0.2-ml PCR tube. Add forward and reverse primers to a final concentration of 0.4 µM each. Add 2.5 µl of 10X PCR amplification buffer (supplied with Taq DNA polymerase). Add dNTPs to a final concentration of 0.2 µM. Add H2O to a final volume of 25 µl (including the volume of Taq DNA polymerase to be added in Step 6). Add 5 units of Taq DNA polymerase. Amplify for 35-40 cycles in a thermal cycler, choosing an appropriate annealing temperature. Transfer 5 µl of the PCR product to another PCR tube. Bring the volume to 25 µl with a newly prepared PCR mix containing [{alpha}-32P]-dCTP (10 µCi/µl, specific activity 3000 Ci/mmol) and fresh dNTPs. The amount of [{alpha}-32P]dCTP should be about 1/100th of the total dCTP in the reaction mixture (i.e., 99/100 cold dCTP, 1/100 radioactive dCTP). Amplify for one additional cycle only. Transfer 10 µl of the hot PCR product obtained in Step 10 into a 1.5-ml microcentrifuge tube. Add 1.5 µl of 10X restriction endonuclease buffer (supplied with the restriction enzyme). Add 10-20 units of a restriction enzyme that cuts the polymorphic restriction site. Digest the sample for 1-2 hours at the recommended temperature (for most enzymes, this will be at 37°C). Add 2.5 µl of 6X DNA loading buffer. Prepare the solution for the polyacrylamide gel. Mix 15 ml of 40% acrylamide/bisacrylamide stock solution, 12 ml of 5X TBE buffer, and 32.5 ml of H2O. Add 50 µl of TEMED and 500 µl of freshly prepared 10% APS. Pour the gel immediately into the vertical electrophoresis tank. Insert the shark's-tooth comb, and clamp it on all sides. Lay the gel flat, and let the matrix polymerize for at least 30 minutes. After polymerization, place the glass plates into the gel apparatus, and add the 1X TBE buffer. Load the samples into the gel, and migrate at 400-800 V for 4-6 hours. After electrophoresis, transfer the gel to a sheet of Whatman 3MM paper, and cover it on one side with plastic wrap. Dry the gel for 45 minutes at 80°C in a gel dryer. Expose the gel to X-ray film in a cassette at room temperature (for 4-16 h), and then develop the film. Alternatively, a phosphorImager can be used to determine the relative intensities of the bands. See Troubleshooting. Use imaging equipment to determine (on the exposed X-ray film or the PhosphorImager picture) the allelic ratio between the undigested and digested PCR products. PCR Amplification to Generate SSCP Polymorphisms (1 day) Place 5-50 ng of template DNA in a 0.2-ml PCR tube. Add forward and reverse primers to a final concentration of 0.4 µM each. Add 2.5 µl of 10X PCR amplification buffer (supplied with the Taq DNA polymerase). Add dNTPs to a final concentration of 0.2 µM. Add H2O to a final volume of 25 µl (including the volume of Taq DNA polymerase to be added). Add 5 units of Taq DNA polymerase. Add 1 µl of [{alpha}-32P]dCTP (10 µCi/µl). Amplify for 35-40 cycles in a thermal cycler. Prepare the solution for the nondenaturing gel. Mix 15 ml of 2X acrylamide solution for SSCP gels, 7.2 ml of 5X TBE buffer, and 37.5 ml of H2O. Add 40 µl of TEMED and 400 µl of freshly prepared 10% APS. TBE will be at a final concentration of 0.6X. Pour the gel immediately into a DNA sequencing gel apparatus. Insert the shark's-tooth comb with teeth pointing upward to form a single well the width of the gel, and clamp on all sides. Lay the gel flat, and let the matrix polymerize for at least 30 minutes. After polymerization, remove the clamps and comb. Reinsert the shark's-tooth comb with teeth pointing downward to form multiple wells. Place the gel into the sequencing gel apparatus, and add 0.6X TBE. To 2 µl of each PCR product, add 8 µl of SSCP loading dye. Denature the samples for 5 minutes at 95°C, then place them on ice. In most cases SSCP separates 150- to 300-bp single-stranded DNA fragments with one or more nucleotide differences. However, the migration of single-stranded fragments in the gel is strongly temperature-dependent. Ideally, therefore, the PCR samples to be compared should be run on the same gel. Following gel electrophoresis, lay the gel on a sheet of Whatman 3MM paper, and cover it on one side with plastic wrap. Dry the gel for 45 minutes at 80°C in a gel dryer. Expose the gel to X-ray film for 4-16 hours at room temperature. A PhosphorImager may be used to determine the relative intensities of the bands. Figure 1 provides an example of results using SSCP. Quantitative PCR Analysis of Precipitated Chromatin Real-Time PCR Amplification (2-3 h) For an example of a real-time PCR protocol, see Real-Time PCR. Run amplifications in triplicate to control for PCR variations. Duplex PCR Amplification (3-4 h) Place 20-50 ng of template DNA in a 0.2-ml PCR tube. Add forward and reverse primers to a final concentration of 0.4 µM each. Add forward and reverse primers for the internal control region (e.g., the actin gene). Add 2.5 µl of 10X PCR amplification buffer (supplied with Taq DNA polymerase). Add dNTPs to a final concentration of 0.2 µM. Add 18.4 µl of H2O. Add 5 units of Taq DNA polymerase. Amplify for 30-35 cycles in a thermal cycler. Take 5 µl of PCR product and add 2 µl of 6X DNA loading buffer. Adjust the volume to 12 µl with H2O. Load the samples onto a 1% (w/v) agarose gel (~10-15 cm in length) in 1X TBE. Load one of the lanes with 1 µg of 100-bp DNA stepladder. Migrate at 2-3 V/cm for 1-2 hours. Stain the gel for 30 minutes in a tray with 500 ml of H2O to which 20 µg of ethidium bromide has been added. Take a photograph of the gel, and check the size of the two different PCR products. Additionally, it is advisable to check that saturation of the amplification reaction (i.e., after 30-35 cycles of PCR) does not change the ratio between the two PCR products. For a semiquantitative test, quantify bands with ImageQuant (Amersham). Calculate the ratio between the different PCR products. To work out precisely the ratio between the two different PCR products, amplification should be performed by adding radioactive [{alpha}-32P]dCTP to the reaction mixture (1/100th of total dCTP), precisely as described in Steps 23-30 of the above-described SSCP analysis. Subsequent electrophoresis is through a standard polyacrylamide gel, as described in Steps 16-22 above. |
|
ACKNOWLEDGEMENTS Our laboratory acknowledges grant funding from the CNRS, the Association pour la Recherche sur le Cancer (ARC), and the ESF EuroCORES Programme EuroSTELLS. TROUBLESHOOTING Problem: Absence or poor restriction enzyme digestion of PCR products. [Step 21] Solution: Some restriction endonucleases do not digest unpurified PCR products, even after addition of the appropriate buffer. Check carefully whether the PCR buffer is compatible with the chosen restriction enzyme. If not, the PCR product should be purified first, following standard procedures, before digestion with the restriction enzyme. Problem: The input chromatin is enriched for one of the two alleles. [Steps 21 or 37] Solution: Try different MNase digestion conditions (shorter/longer digestions) for fractionating the chromatin (see Chromatin Immunoprecipitation on Unfixed Chromatin from Cells and Tissues to Analyze Histone Modifications). Problem: Absence of differential migration of each single strand upon SSCP electrophoresis. [Step 37] Solutions: * Try migration in 1X TBE. * Increase the concentration of acrylamide to 0.7X (Step 31). Problem: Weak or inconsistent separation of single-stranded DNA fragments upon SSCP electrophoresis. [Step 37] Solutions: * To faithfully reproduce the migration of SSCP polymorphisms, it is essential to always run samples at the same temperature (e.g., at room temperature). * SSCP is more efficient for DNA with a relatively high G+C content. SSCP analysis of fragments with a lower G+C content can be enhanced by electrophoresis at 4°C. * Instead of adding radioactive [{alpha}-32P]dCTP to the PCR reactions for SSCP analysis, the PCR primers (forward and reverse) may be radioactively end-labeled by using T4 polynucleotide kinase and [{gamma}-32P]dATP. DISCUSSION For allelic studies on precipitated chromatin fractions, it is essential to include DNA from the input chromatin and from a precipitation with an unrelated control antiserum (see Chromatin Immunoprecipitation on Unfixed Chromatin from Cells and Tissues to Analyze Histone Modifications). This allows verifying whether the different alleles are equally represented in input chromatin, and whether nonspecific background precipitation occurs at one allele preferentially. Also for quantifying precipitated chromatin fractions, it is essential to include DNA from a precipitation with an unrelated control antiserum. This will indicate how much chromatin is brought down nonspecifically at the locus of interest (background precipitation level). REFERENCES Gregory, R.I., Randall, T.E., Johnson, C.A., Khosla, S., Hatada, I., O'Neill, L.P., Turner, B.M., and Feil, R. 2001. DNA methylation is linked to deacetylation of histone H3, but not H4, on the imprinted genes Snrpn and U2af1-rs1. Mol. Cell. Biol. 21: 5426-5436. Noma, K.-I., Allis, C.D., and Grewal, S.I.S. 2001. Transitions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science 293: 1150-1155. Uejima, H., Lee, M.P., Cui, H., and Feinberg, A.P. 2000. Hot-stop PCR: A simple and general assay for linear quantitation of allele ratios. Nat. Genet. 25: 375-376. |
→如果您认为本词条还有待完善,请 编辑词条
词条内容仅供参考,如果您需要解决具体问题
(尤其在法律、医学等领域),建议您咨询相关领域专业人士。
0
收藏到: