|
从头预测模型的基本思想 在既没有已知结构的同源蛋白质、也没有已知结构的远程同源蛋白质的情况下,上述两种蛋白质结构预测的方法都不能用,这时只能采用从头预测方法(Abinitio),即(直接)仅仅根据序列本身来预测其结构。在1994年之前,还没有一个从头算方法能够预测蛋白质的空间结构。从那以后,人们陆续提出一些方法,表明了今后进一步研究可能的方向。有些研究小组运用距离几何方法得到了非常有希望的结果。将简化的力场与动态优化策略相结合,虽然得到的结果不算太精确,但很有意义,表明这样的工作非常有希望突破。 从头预测方法一般由下列3个部分组成:(1)一种蛋白质几何的表示方法:由于表示和处理所有原子和溶剂环境的计算开销非常大,因此需要对蛋白质和溶剂的表示形式作近似处理,例如,使用一个或少数几个原子代表一个氨基酸残基;(2)一种能量函数及其参数,或者一个合理的构象得分函数,以便计算各种构象的能量。通过对已知结构的蛋白质进行统计分析,可以确定蛋白质构象能量函数中的各个参数或者得分函数;(3)一种构象空间搜索技术:必须选择一个优化方法,以便对构象空间进行快速搜索,迅速找到与某一全局最小能量相对应的构象。其中,构象空间搜索和能量函数的建立是从头预测方法的关键。 蛋白质构象的网格模型 限制蛋白骨架构象中可采取的自由度是在模拟过程中简化蛋白质的一种方法,其中一种限制是α碳原子只允许位于二维或三维格子(网格)的位置上。这种简化方法大大减少了一个蛋白质可以采取的构象数目。于是,对于一个中等大小的多肽链,我们可以对它的构象空间进行穷举搜索,直到找到能量全局最小的构象。而对于比较长的多肽链,简化的格子模型可以使非穷尽的搜索方法对所有可能的构象进行较大比例的取样,因此可以比较准确地估计出能量全局最小的构象。 H-P[疏水(hydrophobic)-极性(polar)]模型是研究得最成熟的一种简单网格模型。H-P模型用一个固定半径的原子来表示蛋白质中每个氨基酸残基,从而进一步简化蛋白质结构。在这种表示方法中,原子被分为两种类型:疏水原子和极性原子。如图:
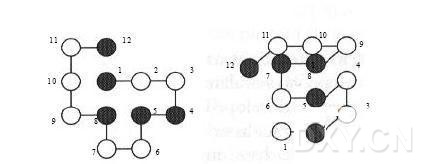
一段较短的用二维和三维H-P模型表示的多肽链 左为二维图,右为三位图(疏水残基表示为黑色,极性残基表示为白色) 按照惯例,N端的氨基酸位于坐标系统的原点,第二个氨基酸残基就位于坐标的(1,0)或(1,0,0)处。通常我们认为疏水作用力是使蛋白质折叠成一个紧密球状结构的几种基础力之一。大多数蛋白质的天然结构都有一个疏水核心和一个与溶液相接触的表面,疏水核心中掩藏了疏水残基,使得它们与溶液相隔离,而与溶液相接触的表面大多或者全部由极性残基和带电残基组成。将蛋白质折叠成一个紧密结构以帮助疏水残基与溶液相分离的过程通常称为疏水折叠。膜蛋白却明显不同,这种蛋白具有一个或多个嵌入细胞膜的跨膜区,这些跨膜区的结构主要是螺旋结构。由于细胞膜大多由疏水的碳原子和氢原子组成,因此这些“表面”的螺旋结构实际上是与水分子分离的,它们大多由疏水氨基酸组成。 H-P模型是基于疏水残基之间的接触来进行打分的。为了评价H-P模型中一个特定的构象,我们要计算出网格中H和H接触的数目。在这里,除了多肽链一级结构中相邻的H和H接触外(由于多肽链一级结构中相邻的H和H接触在每一个可能的构象中都存在,因此为了简单起见这些H和H接触就被去除),其它每一个H和H的接触对能量的贡献都设为-1。最优的构象就是所有可能的构象中具有最多H和H接触的那个构象。一般来说,要获得最大的H和H接触的数目通常需要先形成一个疏水核心,这个疏水核心必须含有尽可能多的H残基,同时要将P残基转移至多肽链的表面。上图中的二维和三维构象的得分都是-3。 有了网格模型及构象能量计算方法,下一个任务就是搜索能量全局最小的构象。在设计搜索算法时,一个主要问题就是如何表示一个特定的构象。一个最简单的方法就是将第一个残基放在网格的(0,0)或(0,0,0)格点上,然后描述前面一个残基到下一个残基的移动方向。二维模型运用这种绝对方向表示法时,每一个位置上可选择的方向包括上、右、左和下(U、R、L、D);而对于三维模型,每一个位置上可选择的方向包括上、右、左、下、后和前(U、R、L、D、B、F)。通过这种绝对方向表示法,可以将上图中的二维构象表示成(R,R,D,L,D,L,U,L,U,U,R),而三维构象可以表示成(R,B,U,F,L,U,R,B,L,L,F)。相对方向表示法则利用每个氨基酸残基主链的转动方向来表示每个位置上的残基的方向,这种方法能够减少每个位置上可选择的方向数。这种情况下,对一个二维正方形的网格模型,第二个残基以后的每个残基位置上可选择的方向有三个,左、右和前(通常表示为L、R和F);对一个三维正方体的网格模型,每个残基位置上可选择的方向有左、右、前、上和下(L、R、F、U、D)。在这种表示方法中,我们不但要清楚当前的位置,同时还要清楚当前残基“面对”的方向。对于二维模型,第一个残基位于网格的(0,0)位上,它所面对的方向为右。也就是说,如果第一个移动方向是F,那么第二个残基就应该位于网格的(1,0)位上。因此,上图中的二维构象用相对方向表示法可表示为(F,F,R,R,L,R,R,L,R,F,R)。对于三维模型,第一个残基位于网格的(0,0,0)位上,它所面对的方向为右。当我们沿着多肽链移动时,我们不但必须清楚当前残基面对的方向,同时还要清楚当前哪个方向应该看作是“上”。利用这种表示方法,上图中的三维构象可以表示为(F,L,U,U,R,U,U,L,L,F,L)。使用上面两种基于方向的表示方法时,我们会遇到的一个关键问题就是一些构象中两个残基会出现在同一个位置上。比如,一个二维构象用相对(基于主链的转动)表示法表示时,如果它的起始四个残基表示为(L,L,L,L),那么这个构象就会有两个残基位于原点(0,0)上,从而导致残基碰撞(bump),或者说原子空间碰撞。在构象搜索时如果出现这种空间碰撞,我们可以采用多种方法来处理。最简单的一种方法就是为每一个具有碰撞的构象分配一个非常高的能量值。由于搜索算法是寻找低能量构象的,因此具有碰撞的构象在搜索时会被很快地剔除。不过,有些构象如果能够解决碰撞问题,它的能量就会比较小,因此这些构象可能会是有效构象。但是,如果采用上面的方法解决碰撞问题的话,搜索过程中就会把这些有效构象去除掉。其它处理碰撞的方法包括在为构象打分之前先利用局部优化方法来解决碰撞,另外也可以使用其他在构象搜索过程中不会产生碰撞的表示法。优先排序表示法就是一种在构象搜索过程中不会产生碰撞的表示法。在优先排序法中,每个残基对应的方向并不是某一个方向,而是所有可能的方向的排列。比如,在二维模型中,某一个残基对应的方向可能会是{L,F,R}。{L,F,R}表示这个残基最可能对应的方向是左;但是,如果残基移向左侧构象中会出现碰撞,这时我们就会为这个残基选择下一个比较有可能的方向,即向前,最后一个可选择的方向为向右。使用这种表示法来表示构象,在有些构象中仍然会出现碰撞(当向所有方向的移动都会导致碰撞时),但这种表示方法中出现碰撞的频率比用绝对方向表示法时出现碰撞的频率要小很多。将优先排序表示法和局部构象搜索方法结合起来,我们就可以设计出构象中绝对不会出现碰撞情况的表示法。 |
|
H-P模型是基于三种简化的,即蛋白质中各个氨基酸残基的α碳原子都位于二维网格或三维网格的格点上,疏水作用是蛋白折叠中唯一的重要因素,同时通过计算疏水残基接触的数目代替构象的能量计算。虽然这样的处理非常简单,但是,通过H-P模型的计算分析,能够发现蛋白质折叠的一些机制。 如果在蛋白质模型中取消氨基酸定位于网格点的限制,那么蛋白模型就可以更真实地模拟出蛋白的实际构象。去网格模型的误差通常用预测构象和实际构象中α碳原子的均方根偏差(RMSD)来计算。α碳原子的RMSD是指当预测构象和实际构象重叠在一起时,两种构象中每个α碳原子位置的Euclidean平方距离的总和。 随着蛋白模型与实际情况越来越相符,模型的复杂性也越来越大。去网格蛋白折叠模型可以只考虑α碳原子,也可以考虑所有的骨架原子,甚至可以考虑所有的骨架原子和侧链原子。假如在模型中考虑侧链的话,那么侧链可以表示成刚性侧链、半柔性侧链和完全柔性侧链。对于刚性侧链,我们已经在X射线结晶结构中得到了这些侧链的构象,X射线结晶结构中每种氨基酸出现最多的构象就被看作这种氨基酸的刚性侧链采取的构象。对于半柔性侧链,我们也是利用类似的经验性方法得到它的构象。从一系列X射线结构中可以得到侧链的多种构象,对这些构象进行分组,形状类似的为一组,这种方法中排除了那些不经常出现的构象,这也减少了搜索的复杂度。 能量函数及优化 除了要考虑疏水作用,蛋白折叠的能量函数中还要考虑到氢键、二硫桥的形成、静电作用、范德华力以及溶剂作用。由于这些力中每一个力的相对作用还很难通过实验来计算,因此寻找一个合适的蛋白折叠复合能量函数仍然是一个研究热点。我们可以通过理论方法,针对范德华力、氢键、溶剂、静电和其它力对一个已折叠蛋白总体稳定性的相对作用来建立能量函数。它的目标是得到一个近似的能量函数或者力场,那些已知结构的蛋白质结晶构象在这个能量函数中处于一个最小能量的状态。如何寻找一些可行的能量函数,本质上是分子力学的问题。而且,科学家确实已经设计出了许多有效的能量函数。 分子力学方法假设正确的蛋白质折叠对应于最低能量的构象。分子力学势能是原子坐标的函数,其极小值对应于原子体系的局部能量最小点。势能函数由多项组成,包括成键作用和非成键作用。成键作用项分为化学键的伸缩能(键长)、弯曲能(键角)和扭转能(二面角),非成键作用包括范德华力、静电力、氢键等。分子力学中的势能参数有各种来源,包括从头算和半经验量子化学计算结果、氨基酸和小分子的实验观察结果等。 对于能量的优化有多种方法。常用的方法是梯度下降法,其中最陡下降法是一种简单的优化算法。在最低能量搜索过程中,最陡下降法反复对能量函数进行微分,计算梯度,每次沿能量下降最多的方向前进。当搜索位置离能量极小点比较远时,用这种方法可以迅速向极小点靠近,但接近极小点时,会产生振荡,收敛速度慢。另一种基于梯度的方法是共轭梯度法,其计算与最陡下降法一样,但是在选择搜索方向时,不仅考虑当前的梯度,还要考虑原来的搜索方向,经过综合决定下一步搜索方向。共轭梯度法收敛的速度快,但是更容易陷入能量局部极小点。 牛顿-拉普森方法是另一类能量优化方法。梯度方法在计算时使用的是一阶微分,而牛顿-拉普森方法除使用一阶微分外,还计算二阶微分,利用一阶微分确定搜索方向,用二阶微分确定沿梯度在什么地方改变方向。应用该方法能够迅速收敛,但是计算量非常大。也可以通过分子动力学来寻找具有局部最低能量的构象。分子动力学利用牛顿力学的基本原理,通过求解运动方程得到所有原子的运动轨迹,并根据轨迹计算各种性质。分子动力学的优势在于能够跨过较大的势垒,获得低能量的构象。在蒙特卡罗和其它理论、实验方法的支持下,分子动力学技术作为改进的模型,在搜索过程中能够避免陷入局部能量极小点。分子动力学另外一个特点是可以模拟蛋白质折叠的过程,从而深入了解蛋白质折叠的规律。 蒙特卡罗是一种随机采样的方法,通过该方法可以期望找到非常接近于全局能量最优的构象。也有用模拟退火方法、遗传算法等进行蛋白质构象搜索和结构预测。 然而,要确保找到全局最低能量的构象,必须进行全面搜索,以一定步长搜索整个构象空间,从而寻找能量最低点。由于搜索的是整个构象空间,所以最终找到的是全局最小点。但是对于生物大分子来讲搜索空间太大,在实际应用中不可行,只能处理很小的蛋白质。即使对搜索空间进行约束,如只允许我们感兴趣的氨基酸和连接两个残基的二面角发生变化,计算量仍然是个问题。对构象空间的进一步简化也只能处理比较小的蛋白质。 虽然利用引起蛋白质折叠的物理力学以及能量函数对蛋白质进行建模有一定实际意义,但是这种从头开始预测蛋白质结构的方法由于种种原因往往得不到令人满意的结果。首先,到目前为止,我们还没有完全了解究竟是哪些力决定了蛋白质的折叠过程,同时这些力之间又是如何相互作用的。即使有了一些力场,但是,力场参数不精确。其次,这种方法需要考虑蛋白质中所有原子之间以及所有原子与周围溶剂之间的相互作用。对于实际大小的多肽,由于计算量太大,这种方法其实并不可行。实际上,也没有对溶剂处理的好方法。再一方面,构象搜索过程容易陷入局部能量极小点,而且自然折叠的蛋白质结构与一般蛋白质构象之间的能量差值比较小,因此,通过计算发现蛋白质的自然折叠结构非常困难。 对于从头开始的方法,另外一种变化方法就是根据一些已知结构的蛋白质构象为一个未知结构的蛋白设计一个经验性的伪能量函数。通常,为得到这种经验性的能量函数表达式,我们首先要选择一系列已知结构的蛋白质,然后对于每一个氨基酸,分析在三维空间上与其相邻的氨基酸。于是,我们可以根据不同氨基酸的相对位置得到一个得分矩阵。例如,得分矩阵中会记录所有丝氨酸残基和苏氨酸残基的距离小于3.6的数目。对一个假定的蛋白质构象,为了估计出它的经验性能量,必须考虑这个蛋白中每个残基的相邻残基。对于那些在样本库中经常出现的局部构象,它们的能量得分会比较小,而对于那些在样本库中不经常出现的局部构象,它们的得分则比较高。如果一个构象的得分比较高的话,这个构象就不太稳定。例如,假如一个特定的丝氨酸残基在6的距离内有三个相邻的残基,即天冬氨酸、组氨酸和谷氨酸,并且得分矩阵显示天冬氨酸、组氨酸和谷氨酸在蛋白结构样本库中经常与丝氨酸相邻,那么这个丝氨酸残基的能量得分就比较低。但是,假如得分矩阵显示丝氨酸和谷氨酸很少相邻,那么这个丝氨酸残基的经验性能量值就比较高。将蛋白质中所有残基的局部能量值累加,就得到这个蛋白质基于经验的全局能量值。实际上,这种经验性能量函数只对那些与已知蛋白质的结构相似的构象赋予比较低的能量值,而对那些新出现的构象或者不经常出现的构象,这种能量函数给出的能量值则比较高。 |
→如果您认为本词条还有待完善,请 编辑词条
词条内容仅供参考,如果您需要解决具体问题
(尤其在法律、医学等领域),建议您咨询相关领域专业人士。
1
收藏到: