|
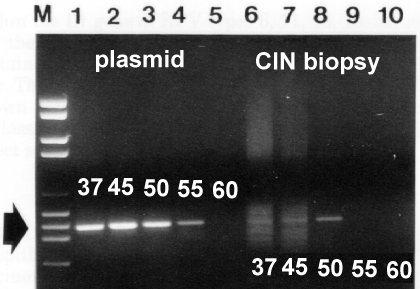
The specific complementary ass℃iation due to hydrogen bonding of single-stranded nucleic acids is referred to as "annealing": two complementary sequences will form hydrogen bonds between their complementary bases (G to C, and A to T or U) and form a stable double-stranded, anti-parallel "hybrid" molecule. One may make nucleic acid (NA) single-stranded for the purpose of annealing - if it is not single-stranded already, like most RNA viruses - by heating it to a point above the "melting temperature" of the double- or partially-double-stranded form, and then flash-cooling it: this ensures the "denatured" or separated strands do not re-anneal. Additionally, if the NA is heated in buffers of ionic strength lower than 150mM NaCl, the melting temperature is generally less than 100℃ - which is why PCR works with denaturing temperatures of 91-97℃. A more detailed treatment of annealing / hybridisation is given in an accompanying page, together with explanations of calculations of complexity, conditions for annealing / hybridisation, etc. Taq polymerase is given as having a half-life of 30 min at 95℃, which is partly why one should not do more than about 30 amplification cycles: however, it is possible to reduce the denaturation temperature after about 10 rounds of amplification, as the mean length of target DNA is decreased: for templates of 300bp or less, denaturation temperature may be reduced to as low as 88℃ for 50% (G+C) templates (Yap and McGee, 1991), which means one may do as many as 40 cycles without much decrease in enzyme efficiency. “Time at temperature” is the main reason for denaturation / loss of activity of Taq: thus, if one reduces this, one will increase the number of cycles that are possible, whether the temperature is reduced or not. Normally the denaturation time is 1 min at 94℃: it is possible, for short template sequences, to reduce this to 30 sec or less. Increase in denaturation temperature and decrease in time may also work: Innis and Gelfand (1990) recommend 96℃ for 15 sec. Annealing Temperature and Primer Design Primer length and sequence are of critical importance in designing the parameters of a successful amplification: the melting temperature of a NA duplex increases both with its length, and with increasing (G+C) content: a simple formula for calculation of the Tm is Tm = 4(G + C) + 2(A + T)℃. Thus, the annealing temperature chosen for a PCR depends directly on length and composition of the primer(s)。 One should aim at using an annealing temperature (Ta) about 5℃ below the lowest Tm of ther pair of primers to be used (Innis and Gelfand, 1990)。 A more rigorous treatment of Ta is given by Rychlik et al. (1990): they maintain that if the Ta is increased by 1℃ every other cycle, specificity of amplification and yield of products <1kb in length are both increased. One consequence of having too low a Ta is that one or both primers will anneal to sequences other than the true target, as internal single-base mismatches or partial annealing may be tolerated: this is fine if one wishes to amplify similar or related targets; however, it can lead to “non-specific” amplification and consequent reduction in yield of the desired product, if the 3'-most base is paired with a target. A consequence of too high a Ta is that too little product will be made, as the likelihood of primer annealing is reduced; another and important consideration is that a pair of primers with very different Tas may never give appreciable yields of a unique product, and may also result in inadvertent “asymmetric” or single-strand amplification of the most efficiently primed product strand. Annealing does not take long: most primers will anneal efficiently in 30 sec or less, unless the Ta is too close to the Tm, or unless they are unusually long. An illustration of the effect of annealing temperature on the specificity and on the yield of amplification of Human papillomavirus type 16 (HPV-16) is given below (Williamson and Rybicki, 1991: J Med Virol 33: 165-171)。
Plasmid and biopsy sample DNA templates were amplified at different annealing temperatures as shown: note that while plasmid is amplified from 37 to 55℃, HPV DNA is only specifically amplified at 50℃. |
|
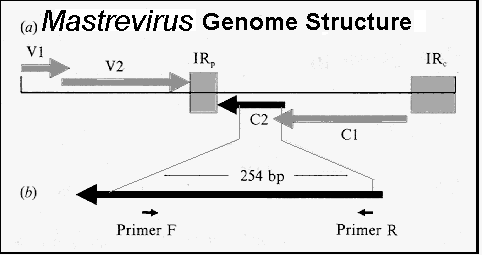
Primer Length The optimum length of a primer depends upon its (A+T) content, and the Tm of its partner if one runs the risk of having problems such as described above. Apart from the Tm, a prime consideration is that the primers should be complex enough so that the likelihood of annealing to sequences other than the chosen target is very low. For example, there is a 1/4 chance (4-1) of finding an A, G, C or T in any given DNA sequence; there is a 1/16 chance (4-2) of finding any dinucleotide sequence (eg. AG); a 1/256 chance of finding a given 4-base sequence. Thus, a sixteen base sequence will statistically be present only once in every 416 bases (=4 294 967 296, or 4 billion): this is about the size of the human or maize genome, and 1000x greater than the genome size of E. coli. Thus, the association of a greater-than-17-base oligonucleotide with its target sequence is an extremely sequence-specific process, far more so than the specificity of monoclonal antibodies in binding to specific antigenic determinants. Consequently, 17-mer or longer primers are routinely used for amplification from genomic DNA of animals and plants. Too long a primer length may mean that even high annealing temperatures are not enough to prevent mismatch pairing and non-specific priming. Degenerate Primers For amplification of cognate sequences from different organisms, or for “evolutionary PCR”, one may increase the chances of getting product by designing “degenerate” primers: these would in fact be a set of primers which have a number of options at several positions in the sequence so as to allow annealing to and amplification of a variety of related sequences. For example, Compton (1990) describes using 14-mer primer sets with 4 and 5 degeneracies as forward and reverse primers, respectively, for the amplification of glycoprotein B (gB) from related herpesviruses. The reverse primer sequence was as follows: TCGAATTCNCCYAAYTGNCCNT where Y = T + C, and N = A + G + C + T, and the 8-base 5'-terminal extension comprises a EcoRI site (underlined) and flanking spacer to ensure the restriction enzyme can cut the product (the New England Biolabs catalogue gives a good list of which enzymes require how long a flanking sequence in order to cut stub ends)。 Degeneracies obviously reduce the specificity of the primer(s), meaning mismatch opportunities are greater, and background noise increases; also, increased degeneracy means concentration of the individual primers decreases; thus, greater than 512-fold degeneracy should be avoided. However, I have used primers with as high as 256- and 1024-fold degeneracy for the successful amplification and subsequent direct sequencing of a wide range of Mastreviruses against a background of maize genomic DNA (Rybicki and Hughes, 1990).
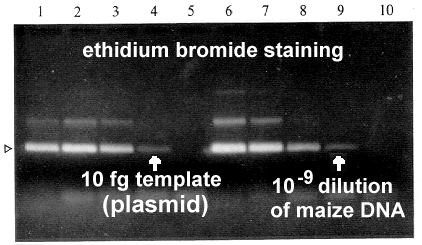
Primer sequences were derived from multiple sequence alignments; the mismatch positions were used as 4-base degeneracies for the primers (shown as stars; 5 in F and 4 in R), as shown above. Despite their degeneracy, the primers could be used to amplify a 250 bp sequence from viruses differing in sequence by as much as 50% over the target sequence, and 60% overall. They could also be used to very sensitively detect the presence of Maize streak virus DNA against a background of maize genomic DNA, at dilutions as low as 1/109 infected sap / healthy sap (see below)。
|
|
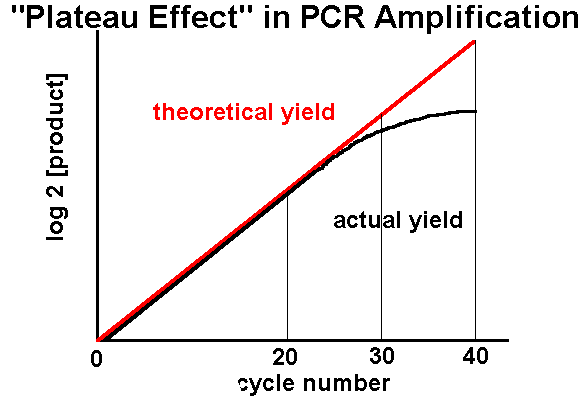
Some groups use deoxyinosine (dI) at degenerate positions rather than use mixed oligos: this base-pairs with any other base, effectively giving a four-fold degeneracy at any postion in the oligo where it is present. This lessens problems to do with depletion of specific single oligos in a highly degenerate mixture, but may result in too high a degeneracy where there are 4 or more dIs in an oligo. Elongation Temperature and Time This is normally 70 - 72oC, for 0.5 - 3 min. Taq actually has a specific activity at 37oC which is very close to that of the Klenow fragment of E coli DNA polymerase I, which accounts for the apparent paradox which results when one tries to understand how primers which anneal at an optimum temperature can then be elongated at a considerably higher temperature - the answer is that elongation occurs from the moment of annealing, even if this is transient, which results in considerably greater stability. At around 70oC the activity is optimal, and primer extension occurs at up to 100 bases/sec. About 1 min is sufficient for reliable amplification of 2kb sequences (Innis and Gelfand, 1990)。 Longer products require longer times: 3 min is a good bet for 3kb and longer products. Longer times may also be helpful in later cycles when product concentration exceeds enzyme concentration (>1nM), and when dNTP and / or primer depletion may become limiting. Reaction Buffer Recommended buffers generally contain : 10-50mM Tris-HCl pH 8.3, up to 50mM KCl, 1.5mM or higher MgCl2, primers 0.2 - 1uM each primer, 50 - 200uM each dNTP, gelatin or BSA to 100ug/ml, and/or non-ionic detergents such as Tween-20 or Nonidet P-40 or Triton X-100 (0.05 - 0.10% v/v) (Innis and Gelfand, 1990)。 Modern formulations may differ considerably, however - they are also generally proprietary. PCR is supposed to work well in reverse transcriptase buffer, and vice-versa, meaning 1-tube protocols (with cDNA synthesis and subsequent PCR) are possible (Krawetz et al., 19xx; Fuqua et al., 1990)。 Higher than 50mM KCl or NaCl inhibits Taq, but some is necessary to facilitate primer annealing. [Mg2+] affects primer annealing; Tm of template, product and primer-template associations; product specificity; enzyme activity and fidelity. Taq requires free Mg2+, so allowances should be made for dNTPs, primers and template, all of which chelate and sequester the cation; of these, dNTPs are the most concentrated, so [Mg2+] should be 0.5 - 2.5mM greater than [dNTP]. A titration should be performed with varying [Mg2+] with all new template-primer combinations, as these can differ markedly in their requirements, even under the same conditions of concentrations and cycling times/temperatures. Some enzymes do not need added protein, others are dependent on it. Some enzymes work markedly better in the presence of detergent, probably because it prevents the natural tendency of the enzyme to aggregate. Primer concentrations should not go above 1uM unless there is a high degree of degeneracy; 0.2uM is sufficient for homologous primers. Nucleotide concentration need not be above 50uM each: long products may require more, however. Cycle Number The number of amplification cycles necessary to produce a band visible on a gel depends largely on the starting concentration of the target DNA: Innis and Gelfand (1990) recommend from 40 - 45 cycles to amplify 50 target molecules, and 25 - 30 to amplify 3x105 molecules to the same concentration. This non-proportionality is due to a so-called plateau effect, which is the attenuation in the exponential rate of product accumulation in late stages of a PCR, when product reaches 0.3 - 1.0 nM. This may be caused by degradation of reactants (dNTPs, enzyme); reactant depletion (primers, dNTPs - former a problem with short products, latter for long products); end-product inhibition (pyrophosphate formation); competition for reactants by non-specific products; competition for primer binding by re-annealing of concentrated (10nM) product (Innis and Gelfand, 1990)。
|
|
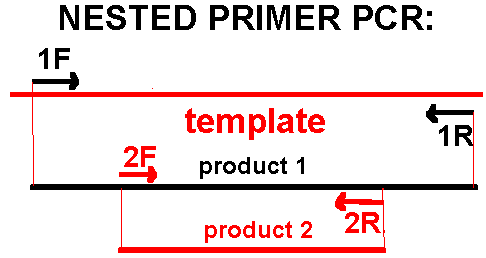
If desired product is not made in 30 cycles, take a small sample (1ul) of the amplified mix and re-amplify 20-30x in a new reaction mix rather than extending the run to more cycles: in some cases where template concentration is limiting, this can give good product where extension of cycling to 40x or more does not. A variant of this is nested primer PCR: PCR amplification is performed with one set of primers, then some product is taken - with or without removal of reagents - for re-amplification with an internally-situated, “nested” set of primers. This process adds another level of specificity, meaning that all products non-specifically amplified in the first round will not be amplified in the second. This is illustrated below:
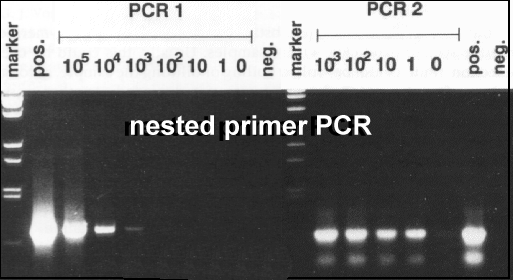
This gel photo shows the effect of nested PCR amplification on the detectability of Chicken anaemia virus (CAV) DNA in a dilution series: the PCR1 just detects 1000 template molecules; PCR2 amplifies 1 template molecule (Soiné C, Watson SK, Rybicki EP, Lucio B, Nordgren RM, Parrish CR, Schat KA (1993) Avian Dis 37: 467-476)。 Labelling of PCR products with digoxygenin-11-dUTP (DIG; Roche) need be done only in 50uM each dNTP, with the dTTP substituted to 35% with DIG-11-dUTP. NOTE: that the product will have a higher MW than the native product! This results in a very well labelled probe which can be extensively re-used, for periods up to 3 years. Helix Destabilisers / Additives With NAs of high (G+C) content, it may be necessary to use harsher denaturation conditions. For example, one may incorporate up to 10% (w or v/v) : in the reaction mix: these additives are presumed to lower the Tm of the target NA, although DMSO at 10% and higher is known to decrease the activity of Taq by up to 50% (Innis and Gelfand, 1990; Gelfand and White, 1990)。 |
|
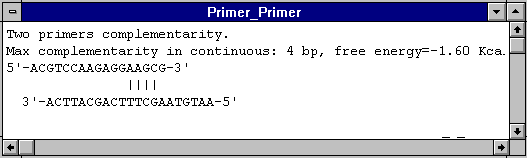
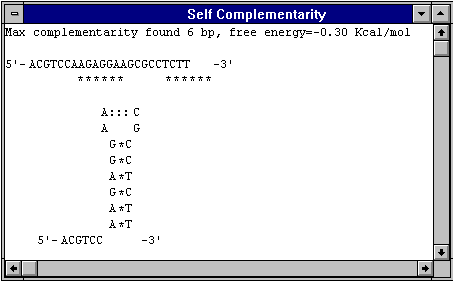
Additives may also be necessary in the amplification of long target sequences: DMSO often helps in amplifying products of >1kb. Formamide can apparently dramatically improve the specificity of PCR (Sarkar et al., 1990), while glycerol improves the amplification of high (G+C) templates (Smith et al., 1990)。 Polyethylene glycol (PEG) may be a useful additive when DNA template concentration is very low: it promotes macromolecular association by solvent exclusion, meaning the pol can find the DNA. cDNA PCR A very useful primer for cDNA synthesis and cDNA PCR comes from a sequencing strategy described by Thweatt et al. (1990): this utilised a mixture of three 21-mer primers consisting of 20 T residues with 3'-terminal A, G or C, respectively, to sequence inside the poly(A) region of cDNA clones of mRNA from eukaryotic origin. I have used it to amplify discrete bands from a variety of poly(A)+ virus RNAs, with only a single specific degenerate primer upstream: the T-primer may anneal anywhere in the poly(A) region, but only molecules which anneal at the beginning of the poly(A) tail, and whose 3'-most base is complementary to the base next to the beginning of the tail, will be extended. eg: 5'-TTTTTTTTTTTTTTTTTTTTTTTTT(A,G,C)-3' works for amplification of Potyvirus RNA, and eukaryotic mRNA A simple set of rules for primer sequence design is as follows (adapted from Innis and Gelfand, 1991): primers should be 17-28 bases in length; base composition should be 50-60% (G+C); primers should end (3‘) in a G or C, or CG or GC: this prevents “breathing” of ends and increases efficiency of priming; Tms between 55-80oC are preferred; runs of three or more Cs or Gs at the 3'-ends of primers may promote mispriming at G or C-rich sequences (because of stability of annealing), and should be avoided; 3'-ends of primers should not be complementary (ie. base pair), as otherwise primer dimers will be synthesised preferentially to any other product; primer self-complementarity (ability to form 2o structures such as hairpins) should be avoided. Examples of inter- and intra-primer complementarity which would result in problems:
Screen shots taken from analyses done using DNAMAN (Lynnon Biosoft, Quebec, Canada)。 |
→如果您认为本词条还有待完善,请 编辑词条
词条内容仅供参考,如果您需要解决具体问题
(尤其在法律、医学等领域),建议您咨询相关领域专业人士。
0
收藏到: