|
摘要:通过PCR,从E.coli K12 Y1090株系中扩增获得glgC基因,插入pUC19的PstI和SacI 位点之间,得到重组质粒pAGP。对所克隆的glgC基因进行全序列测定,结果表明,与已发表的序列相比,有7个核苷酸发生了改变,核苷酸顺序的同源性为99.4 %; 推导的氨基酸序列的同源性为99.2 %, 其中,第296位由赖氨酸变为谷氨酸。通过寡核苷酸介导的定点诱变,将第1007位核苷酸由G变为A,从而使第336位氨基酸由甘氨酸变为天门冬氨酸,将此glgC的突变基因定名为glgC336。经I2-KI染色法鉴定,突变基因的表达可提高细菌中的糖原含量。 关键词:AGPase; E.coli ; PCR;glgC基因;序列分析;定点突变 Cloning And Site-directed Mutagenesis of glgC Gene From E.coli Y1090 Dong Yunzhou Wang Xiaofeng Jia Shirong (Biotechnology Research Center, Chinese Academy of Agricultural Sciences, Beijing 100081) Abstract: ADPglucose pyrophosphorylase(AGPase)is the key enzyme catalyzing the rate-limiting reaction of starch biosynthesis in bacteria and plants. By using genomic DNA of E.coli K12 strain Y 1090 as a template, glgC gene was amplified by polymerase chain reaction(PCR)and cloned into pUC19. The full length of the glgC gene(1296 bp)was sequenced. The nucleotide sequence is 99.4 % homologous to the published glgC gene. The deduced amico acid sequence is 99.2 % homologous to the known sequence. It is noticed that amino acid was changed at the position 296 from lysine to glutamic acid. The nucleotide 1066 was changed from G to A via site-directed mutagenesis, and the amino acid was changed from glycine 336 to aspartic acid accordingly. The mutated gene, named glgC336 that mutated both at the position of 296(Lys→Glu) and 336(Gly →Asp), is able to increase glycogen synthesis in E.coli . Key words: AGPase; E.coli ; glgC gene; PCR; sequence; site-directed mutagenesis 糖原是细菌细胞内碳源及能源的贮藏形式,其作用与淀粉在植物中的作用相同。糖原主要以ADPG途径合成[1]。AGPase (ADPglucose pyrophosphorylase, 腺苷酸二磷酸葡萄糖焦磷酸化酶)是糖原合成中的关键酶[1~3],它催化的反应为ATP+G-1-PADPG+PPi。ADPG是葡萄糖的活化形式,它作为葡糖基的供体,使葡聚糖不断得以延长。大肠杆菌的AGPase是一个同型四聚体,由4个相同的亚基构成,每个亚基的分子量为50 kD,其编码基因为glgC,DNA全序列已测定[4]。Cattaneo等[5]发现了E.coli K12株系中 的一个突变体,其糖原含量比野生型高30%以上。Leung等克隆了该基因,定名为glgC16[2]。Lee(1987)和Kumar等(1989)证明第886位A→G和第1007位G→A的核苷酸双突变是导致AGPase变构调节特性的内因[6,7],这一特性为改良植物的淀粉含量或品质奠定了基础。Stark等(1992)将glgC16基因转入马铃薯,获得了淀粉含量提高的转基因植株,平均含量比对照高35 %, 最高的达60 % [8]。本研究首先对glgC基因进行克隆和定点突变,旨在通过转基因技术,改善我国马铃薯品种淀粉含量偏低(13 %~19 %)的现状, 为食品工业提供优良的原料。 1 材料与方法 1.1 材料 细菌菌株: E.coli Y1090, 来自新加坡国立大学;XLI-Blue为本室保存。质粒:pUC19, pBluescript KS为本室保存;pALTER-1购自Promega公司。工具酶多为Promega、BRL及华美公司产品。PCR引物和定点突变引物由新加坡国立大学分子和细胞生物学研究所合成。DNA手工测序,采用USB公司Sequenase version 2.0 kit。同位素α-32 P-dATP购自亚辉公司;药品、试剂为进口或国产分析纯。 1.2 方法 1)E.coli Y1090染色体DNA的提取参照文献[9]进行。 2)PCR扩增glgC基因,所设计的PCR引物为: 5'-端5'-GACTGCAGGATGGTTAGTTTAGAGAAGAACGATCACTTAATGTTG-3'(45mer) 3'-端 5'-CGAGCTCTTATCGCTCCTGTTTATGCCCTAACTTCCG-3'(37mer)。PCR方法参照分子克隆实验指南[10]。PCR仪为PE9600型,反应程序为94℃ 1 min, 55℃ 1 min, 72℃ 1 min, 共32个循环,循环结束后,72℃ 延伸5min。 3)目的基因的分子克隆与亚克隆按分子克隆实验指南进行[10]。 4)DNA序列分析,基因全序列由中科院微生物所全自动测序仪完成。定点突变DNA片段的手工测序参照分子克隆实验指南进行[10]。 5) 定点突变,按Promega公司Altered sites ® II in vitro mutagenesis kit说明书进行。引物为AKC2:5'-ACAACCGTCGGAAACCAG-3'(18 mer)。 6)目的基因表达产物的生物学活性分析,分别将glgC及其突变基因glgC336克隆到pBluescript KS载体上,转化大肠杆菌XL1-Blue菌,挑取单菌落,在加富Cornberg培养基(0.85 % KH2PO4,1.1 % K2HPO4, 0.6 % Yeast extract, 1 % glucose, 1.5 % Agar)上培养24 h(37℃),然后在菌落上滴加25 L I2-KI溶液(0.1 mol/L I2, 0.3 mol/L KI), 比较菌落棕红色的深浅。 |
|
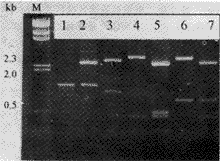
2 结果 2.1 glgC基因的PCR扩增、克隆与酶切鉴定 以E.coli Y1090总DNA为模板,进行PCR扩增,1%agarose 胶电泳鉴定,在1.3 kb处有一条特异性带(图1)。经纯化后的PCR产物与载体pUC19用PstI-SacI双酶切,连接后转化XLI-Blue, 挑选重组子,定名为pAGP(图2),分别选用EcoRI、BamHI、HindIII、SacI、PstI等进行酶切鉴定,都得到了预期大小的DNA片段(图1),初步表明已克隆到glgC基因。
图1 PCR产物及pAGP重组质粒的酶切分析 Fig.1 PCR product and restriction analysis of pAGP M.-HindIII marker;1.PCR product;2.Pst/SacI;3.BamHI/SacI 4.EcorR I;5.EcoRI/PstI;6.HindIII;7.HindIII/SacI
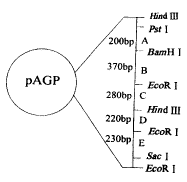
图2pAGP重组质粒图谱 Fig.2 Map of recombinant plasmid pAGP 2.2 glgC基因的序列分析 DNA全自动测序仪测定了全长基因(1296 bp), 对自动测序有疑问的位点进行了二次手工测序。已发表的glgC序列[4]相比,共有7个核苷酸发生了改变,如表1。 表1 克隆的glgC基因与已发表核苷酸序列及其推导的氨基酸序列的比较 Table 1 Comparision of the nucleotide and deduced amino acid sequence between the cloned glgC and previously reported glgC gene
|
|
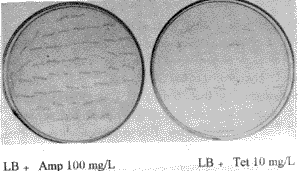
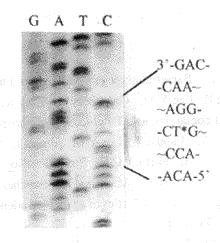
2.3 定点突变 2.3.1 靶序列亚克隆与突变载体pALTER-1 pKA(pBluescript KS/ glgC重组质粒)DNA以HindIII和SacI双酶切,回收片段DE(450 bp), 亚克隆于pBluescript KS载体,定名为pDE。以HindIII和EcoRI双酶切pDE, 回收片段D(220 bp), 将此片段亚克隆于pALTER-1, 得重组质粒pAMD(图3)。 2.3.2 定点突变 定点突变采用Promega公司Altered sites ® II Mutagenesis system。在突变反应中,除加入突变引物AKC2外,还加有Amp repair oligonucleotides 和Tet Knocout oligonucleotides。pALTER-1本身有Tet(四环素)抗性,而没有Amp(氨苄青霉素)抗性,反应过程中加入了上述两个附加引物,从而获得了Amp 抗性而失去了Tet抗性,以此可以汰除大多数非突变体。Amp平板上长出的菌落,重新在Amp 平板和Tet平板上同时划线培养,以计算突变率和挑选克隆。能在Amp 平板上生长而不能在Tet平板上生长的菌落,可初步断定为突变体,突变体可通过测序进一步确定。结果表明,所划的100个菌落,在Amp平板上全部长出,但在Tet平板上只长出19个,突变率为81% (图4)。挑选3个克隆进行测序,结果表明,3个克隆均在预定位置上发生了突变(图5),说明突变反应是成功的。将定点突变基因定名为glgC336。
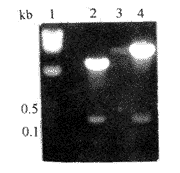
图3 靶序列亚克隆至突载体pALTER-1 Fig3 Subcloning of target fragement to pALTER-1 1λ-HindIII marker;2 pED,EcoR/HindIII;3 pALTER-1,EcoR-1,EcoRI/HindIII;4 pAMD,EcoRI/HindIII
图4 检测突变率 Fig.4 Detection of mutation ratio
图5 测序鉴定靶位点突变 带*者为突变的位点(检测的互补性) Fig.5 Detection of target site mutation by sequencing *indicate mutation site (antisense strand) |
|
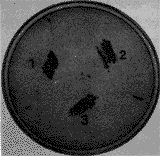
2.4 glgC 和glgC336基因的生物学活性鉴定 将glgC 和glgC336分别克隆于pBluescript KS,获得重组质粒pKA(pBluescript KS / glgC)、pKMA(pBluescript KS / glgC336)。 分别将pBluescript KS、pKA和pKMA转化XLI-Blue , 铺板后培养12 h,在加富Cornberg培养基上划线培养,37℃,24 h; 在菌落上滴加I2-KI溶液,菌落即呈现棕红色,含pKA和pKMA 的宿主菌,其颜色明显深于pBluescript KS宿主菌,而含pKMA 的宿主菌比含pKA 宿主菌的颜色更深(图6)。 根据Chester(1967)和Govons等(1969)的报道:菌落颜色的深浅与它们积累的糖原呈正相关,即颜色越深,糖原含量越高[11,12]。因此,我们可以初步断定,glgC 和glgC336基因表达后,可明显提高宿主菌的糖原含量,且glgC336基因的表达活性高于glgC 基因。
图6 碘-碘化钾染色鉴定菌落的糖原含量 Fig 6 Staining of cell colony with I2-KI 1 含pBluescript KS的宿主菌Host bacterivm contbacterium containing pKA;3 含pKMA的宿主菌Host bacterium containing PKMA 3 讨论 根据前人的研究,我们从E.coli K12的Y1090菌株中通过PCR扩增到了glgC基因,核苷酸序列有7个发生了改变,氨基酸有3个发生了改变,其中第296位氨基酸由Lys→Glu, 这一结果可能有两个原因:(1)PCR扩增时随机突变,Taq酶缺乏校正功能,在PCR过程中,核苷酸参入错误率可达每循环2×10-4个核苷酸。(2)Y1090菌株中该基因已突变。如果是第二种原因,则我们的结果与Kumar等[7]的结果相符,即第296位氨基酸单独突变不会引起该酶活性的改变。 利用定点突变对第1007位核苷酸进行了突变(G→A),将突变后的基因glgC336克隆于pBluescript KS载体上,获得重组质粒pKMA,I2-KI染色结果表明:含glgC336基因的菌落,其颜色比glgC基因的菌落深,说明前者的糖原含量较高,表明第296位和第336位氨基酸同时突变后的AGPase比仅第296位氨基酸突变的AGPase活性增强,这与前人的结果相符[7, 8]。 新加坡国立大学分子和细胞生物学研究所Anil Kush博士为我们合成了PCR引物和定点突变引物;中科院遗传所朱桢先生和陈荣小姐在定点突变实验中给予了大力帮助 ;郭三堆研究员在实验设计中提供了许多宝贵意见,在此一并致谢。 * 863青年基金资助项目 作者简介:董云洲:男,36岁,硕士,副研究员 作者单位:中国农业科学院生物技术研究中心, 北京 100081 参考文献 1 Preiss J. Bacterial glycogen synthesis and its regulation. Ann Rev Microbial, 1984, 38:419~458 2 Leung P, Lee Y M, Greenbery E, Esch K, Boylan S, Preiss J. Cloning and expression of the E.coli glgC gene from a mutant containing an ADPglucose pyrophosphorylase with altered allosteric properties. J of Bacteriol, 1986, 167:82~88 3 Kumar A, Tanaka T, Lee Y M, Preiss J. Biosynthesis of bacterial glycogen. J of Bio Chem, 1988, 263:14634~14639 4 Baecker P A, Furlong C E, Preiss J. Biosynthesis of bacterial glycogen. J of Biol Chem,1983, 258:5084~5088 5 Cattaneo J, Oamotte M, Sigal N, Sanchez-Medina G, Puig J. Genetic studies of E.coli K12 mutants with alterations in glycogenesis and properties of an antered adenosine diphosphate glucose pyrophosphorylase. Biochem Biophys Res, Commun, 1969, 34:694~701 6 Lee Y M, Kumar A , Preiss J. Amino acid sequence of an E.coli ADPglucose synthetase allosteric mutants as deduced from the DNA sequence of the glgC gene. Nucleic Acids Res, 1987, 15: 10603 7 Kumar A, Ghosh P, Lee Y M, Hill M A, Preiss J. Biosynthesis of bacterial glycogen. J of Bio Chem, 1989, 264: 10464~10471 8 Stark D M, Timmerman K P, Barry G F, Preiss J, Kishore G M. Regulation of the amount of starch in plant tissues by ADPglucose pyrophosphorylase. Science, 1992, 258: 287~292 9 李德葆,徐平 . 重组DNA的原理和方法. 浙江: 浙江科技出版社, 1993, 129~130 10 金冬雁,等译. 分子克隆实验指南(第二版). 北京: 科学出版社, 1992, 762~683; 16~50; 602~607 11 Chester V E. Spontaneous mutants of yeast deficient in glycogen. Nature, 1967, 214: 1237~1238 12 Govons S, Vinopal R, Ingraham J, Preiss J. Isolation of mutants of E.coli B altered in their ability to synthesize glycogen. J of Bacteriol, 1969, 97: 970~972 |
→如果您认为本词条还有待完善,请 编辑词条
上一篇DNA extraction from blood-Molecular Biology 下一篇Preparation?of?blunt-end?DNA?fragmen
词条内容仅供参考,如果您需要解决具体问题
(尤其在法律、医学等领域),建议您咨询相关领域专业人士。
0
收藏到: